

Abstract
Systematic variation of the 1,4‐dioxane (dx) concentration during the precipitation of sparingly soluble [MgBr2(dx)2] from ethereal Grignard solutions of RMgBr has allowed the structural investigation of crystallized [R2Mg(dx)n] (n=1, 1.5, 2, and 3), which form during this dioxane method, depending on the bulkiness of R. The numbering of the complexes explored in this study is based on the number n of dioxane molecules per magnesium atom, followed by the substituent R; an apostrophe denotes coordination polymers. The following derivatives were studied by X‐ray crystal‐structure determination and NMR spectroscopy: n=1: [Me2Mg(μ‐dx)]∞ (1′‐Me) and [nPr2Mg(μ‐dx)]∞ (1′‐nPr); n=1.5: [{iPr2Mg(dx)}2(μ‐dx)] (1.5‐iPr), [{oTol2Mg(dx)}2(μ‐dx)] (1.5‐oTol), and [(Me3Si‐C≡C)2Mg(dx)1.5]∞ (1.5′‐C2SiMe3); n=2: [tBu2Mg(dx)2] (2‐tBu) and [oTol2Mg(dx)2] (2‐oTol); n=3: [Ph2Mg(dx)3] (3‐Ph). In the structure types 1′, 1.5, and 2, the magnesium atom exhibits the coordination number 4, whereas pentacoordinate metal atoms are observed in types 3 and 1.5′. The structure type 2′ is realized for [(Ph‐C≡C)2Mg(dx)2]∞ (2′‐C2Ph), [MgCl2(dx)2]∞ (2′‐Cl), and [MgBr2(dx)2]∞ (2′‐Br) with hexacoordinate metal atoms. The solubility of the dioxane adducts in common organic solvents strongly depends on the degree of aggregation with the solubility decreasing from molecular to strand to layer structures.
Keywords: diorganylmagnesium, dioxane method, Grignard reaction, magnesium, Schlenk equilibrium, structure–solubility relationships
Complexities of magnesium! The dioxane method is commonly used to precipitate magnesium halides from Grignard solutions. Elucidation of the structure–solubility relationship has shown that hexacoordinate magnesium atoms and the layer structures of magnesium‐based halides and organometallics (see figure) provoke insolubility in ethereal solutions.
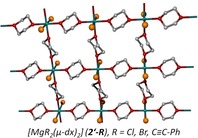
Introduction
Grignard reagents, namely organylmagnesium halides, can be straightforwardly synthesized by the reduction of halohydrocarbons with magnesium turnings (direct synthesis).1 In solution, complex and solvent‐dependent equilibria interconvert mono‐ and oligonuclear species leading to mixtures of RMgX, MgR2, and MgX2; a simplified picture is shown in Scheme 1.
Scheme 1.
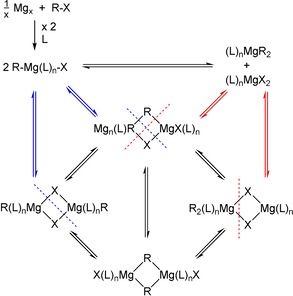
Formation of the Grignard reagent (top) and Schlenk equilibrium (bottom) of organylmagnesium halides interconverting RMgX into homoleptic MgR2 and MgX2 (L is an electroneutral Lewis base such as an ether). Colored dashed lines symbolize deaggregation possibilities leading to the equilibria shown with arrows in the same color.
There has been great interest in halide‐free di(hydrocarbyl)magnesium compounds for several decades.1, 2 Besides the direct synthesis (yielding Grignard reagents), hydromagnesiation of alkenes with activated magnesium hydride3 and the THF method by the precipitation of sparingly soluble Mg(thf)6I2 from RMgI solutions4 have allowed, in special cases, the isolation of MgR2. However, an approved and reliable procedure for the removal of magnesium halides from Grignard solutions, discovered by Schlenk 90 years ago, was realized by the addition of 1,4‐dioxane (dx, the dioxane method), as depicted in Scheme 2.5
Scheme 2.
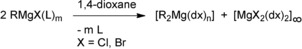
Dioxane method to shift the Schlenk equilibrium of organylmagnesium halides toward soluble [R2Mg(dx)n] and insoluble [MgX2(dx)2]∞ by substitution of the Lewis base L (e.g., diethyl ether or thf) by 1,4‐dioxane (dx).
Diverse observations led to refined procedures for the dioxane method. It proves to be very beneficial to allow the MgX2‐containing precipitate to age overnight at room temperature. The solid is then less voluminous and more compact, which allows a less time‐consuming removal by filtration. During this time, initially trapped MgR2 dissolves, which enhances the yield of diorganylmagnesium. During solvate formation, dioxane competes with the Lewis basic solvent L of the Grignard reaction, the donor strength of the ethers increasing in the order Et2O≪thf≤dx. Consequently, the precipitation of magnesium halides from diethyl ether solutions is nearly quantitative and application of a very small excess of 1,4‐dioxane is sufficient, whereas in THF solution even a large excess of dx still leads to incomplete removal of MgX2. Dioxane molecules can coordinate to metal cations through three different binding modes: The chair conformation enables η1‐terminal as well as μ‐O,O′‐bridging coordination modes, whereas the boat conformation with an η2‐O,O′‐chelating binding mode is as yet unknown in organomagnesium chemistry. The filtrate contains diorganylmagnesium complexes [R2Mg(dx)n], but the use of very bulky R groups can lead to soluble RMgX(dx)n compounds, showing that the Schlenk equilibrium is not always quantitatively shifted toward the homoleptic congeners.6 Ten years ago, the influence of dioxane on the Schlenk equilibrium was studied more explicitly.7 Further examples of dioxane adducts of diorganylmagnesium are known and have been structurally authenticated.8 These compounds are depicted in Scheme 3.
Scheme 3.
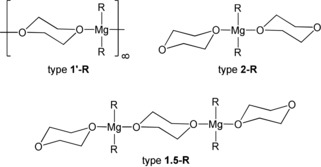
Structural diversity of hitherto known 1,4‐dioxane adducts of diorganylmagnesium complexes authenticated by X‐ray crystal‐structure determinations.
The compounds are labeled considering the number n of 1,4‐dioxane ligands, with an apostrophe symbolizing coordination polymers, followed by the hydrocarbyl group R. Thus, 1′‐Me characterizes [Me2Mg(μ‐dx)]∞. Besides mononuclear complexes of the type [R2Mg(η1‐dx)2] (2‐R with R=Mes,7 2‐MeC2B10H10,9 and C9H7 (indenyl)10), dinuclear congeners [{R2Mg(dx)}2(μ‐dx)] (type 1.5‐R with R=Bz7) have also been investigated. Strand‐like structures such as [R2Mg(μ‐dx)]∞ (type 1′‐R with R=Et,8a CH2 tBu,8b iPr,8c Ph,8d Cy,7 and Cp11) with bridging dioxane ligands are observed in most cases in the crystalline state. The mononuclear silanide complex [{(Me3Si)2MeSi}2Mg(dx)2] of type 2,12 the strand‐like magnesiate [LiMg(CH2SiMe3)3(μ‐dx)(η1‐dx)]∞,13 and the tetranuclear heteroscorpionate complex [{L(MgCH2SiMe3)2}2(μ‐dx)] expand this class of dioxane adducts.14 Furthermore, mononuclear molecular congeners [MgR2] without co‐ligands can be stabilized with very bulky groups such as C(SiMe3)3 and 2,4,6‐tBu3C6H2 (Mes*).15
To systematically investigate the coordination chemistry of dioxane adducts of organomagnesium complexes, we chose simple hydrocarbyl groups without additional heteroatoms. The di(hydrocarbyl)magnesium derivatives were studied in solution and in the crystalline state, depending on the dioxane concentration as well as the size and nature of the hydrocarbyl group R. Thus, we varied the following parameters:
The chain length of aliphatic carbanions (R=Me, Et, nPr, and nBu),
The degree of substitution of aliphatic carbanions (R=Me, Et, iPr, and tBu),
The steric demand of aromatic carbanions (R=Ph, oTol, Mes, and Mes*),
Slim di(alkynyl)magnesium–dioxane adducts (R=C≡C‐Ph, C≡C‐SiMe3).
This selection of organic ligands has allowed us to elucidate the influence of steric pressure and hybridization, because the organic groups R encompass sp3‐ (alkyl), sp2‐ (aryl), and sp‐hybridized (alkynyl) anionic carbon atoms. Furthermore, various molar ratios of MgR2/dx were employed during the crystallization procedures.
Results and Discussion
n‐Alkylmagnesium–dioxane adducts
We first studied the effect of the chain length of alkyl groups on the properties and molecular structures of [R2Mg(dx)n] after precipitation and removal of the magnesium halides. The simplest derivative is dimethylmagnesium.16 Isolation of this compound succeeded by the precipitation of magnesium bromide with 1,4‐dioxane in commercially available solutions of methylmagnesium bromide in THF. After removal of the solvents, the sparingly soluble residue was recrystallized from a mixture of toluene and 1,4‐dioxane to yield colorless and highly pyrophoric crystals of the coordination polymer [Me2Mg(μ‐dx)]∞ (1′‐Me) depicted in Figure 1.
Figure 1.
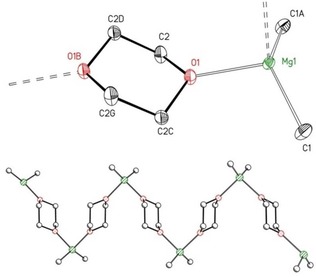
Molecular structure and numbering scheme of [Me2Mg(μ‐dx)]∞ (1′‐Me, top). The ellipsoids represent a probability of 30 %. Hydrogen atoms are not shown for reasons of clarity. The strand structure is depicted at the bottom. Selected bond lengths [pm]: Mg1−C1 212.98(16), 208.88(11); bond angles [°]: C1‐Mg1‐C1A 135.19(9), O1‐Mg1‐O1A 95.04(7), O1‐Mg1‐C1 104.92(3), O1‐Mg1‐C1A 104.92(3).
The crystal structure consists of parallel zigzag chains of MgMe2 units bridged by dioxane ligands with chair conformations. The dx ligands are arranged in a face‐to‐face manner. This arrangement leads to a zigzag chain with a larger amplitude than observed for the coordination polymers of the type [R2Mg(μ‐dx)]∞ with R=Et, Ph, Cy, and iPr. The Mg⋅⋅⋅Mg distance of 889.14(3) pm between the first and third MgMe2 moieties in 1′‐Me is significantly smaller than in other magnesium congeners of this type with values of approximately 1150–1240 pm.17 In all these complexes the magnesium‐bound dx ligands with chair conformations are arranged back‐to‐back (Scheme 4). The nonbonding Mg⋅⋅⋅Mg distance of 690.5(2) pm between neighboring magnesium atoms is determined by the dx ligands and is very similar for all derivatives with a strand structure.18 Due to the larger spacing of the MgR2 units with dioxane ligands arranged back‐to‐back, this is the favored structure for larger R groups.
Scheme 4.
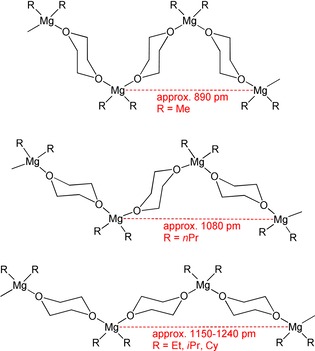
Three possible arrangements of the 1,4‐dioxane bases in coordination polymers of the type [R2Mg(μ‐dx]∞. Top: face‐to‐face; middle: face‐to‐back; bottom: back‐to‐back.
Complex 1′‐Me is only very sparingly soluble in diethyl ether, but readily dissolves in THF and is soluble in warm 1,4‐dioxane. From a pure dioxane solution, again strand‐like 1′‐Me crystallized and a complex containing more dioxane was not observed. The NMR spectra in [D8]THF solution show the resonances for the magnesium‐bound methyl groups at δ(1H)=−1.82 ppm and δ(13C)=−16.5 ppm (1 J C,H=105.6 Hz). In this solvent, the strand structure is deaggregated by the substitution of dx ligands by THF Lewis bases.
The addition of 1,4‐dioxane to an ethereal solution of ethylmagnesium bromide and removal of the precipitated magnesium bromides yielded the known strand structure [Et2Mg(μ‐dx)]∞ (1′‐Et).8a This compound is only soluble in diethyl ether or benzene if dioxane has been added. The strand structure [Et2Mg(μ‐dx)]∞ crystallized again from a solvent mixture of dioxane and diethyl ether (ratio of 5:1).
The dioxane adduct of di(n‐propyl)magnesium is soluble in diethyl ether and this complex crystallized again within several days from a 2.0 m solution of dioxane and diethyl ether (ratio of dx and Et2O of approx. 1:1) at −40 °C to yield colorless crystals of [nPr2Mg(μ‐dx)]∞ (1′‐nPr), as shown in Figure 2.
Figure 2.
Molecular structure and numbering scheme of [nPr2Mg(μ‐dx)]∞ (1′‐nPr, top). The ellipsoids represent a probability of 30 %. Hydrogen atoms have been omitted for the sake of clarity. The strand structure is shown at the bottom. Selected bond lengths [pm]: Mg1−C1 215.20(12), Mg1−C4 215.57(12), Mg1−O1 212.05(9), Mg1−O2 209.31(8); bond angles [°]: C1‐Mg1‐C4 140.64(5), O1‐Mg1‐O2 96.55(4), O1‐Mg1‐C1 102.05(5), O1‐Mg1‐C4 100.01(4), O2‐Mg1‐C1 105.23(4), O2‐Mg1‐C4 104.13(4).
The crystal structure of 1′‐nPr also consists of parallel zigzag chains of (nPr)2Mg moieties bridged by 1,4‐dioxane bases. However, the dioxane ligands show a face‐to‐back arrangement, as depicted in Scheme 4. In comparison with 1′‐Me, this orientation of the dx ligands leads to an elongation of the distance between the first and third magnesium atoms to 1080.96(8) pm. The nonbonding Mg1⋅⋅⋅Mg1A distance between neighboring magnesium atoms is 698.7(1) pm.
Contrary to the shorter n‐alkyl complexes, (nBu)2Mg did not crystallize from a 4.0 m ethereal solution at −40 °C. At −78 °C and upon layering with n‐pentane, an amorphous solid with the composition [nBu2Mg(dx)] precipitated. The longer alkyl groups significantly enhances the solubility in ethereal solvents, thereby causing a deterioration in its crystallization behavior. Probably, the longer alkyl groups do not fit as well between the chains and smaller degrees of aggregation are realized.
1,4‐Dioxane adducts of (Me3−xHxC)2Mg
We next investigated the effect of the degree of substitution on the molecular structures of the dx adducts of (Me3−xHxC)2Mg (x=3: Me; 2: Et; 1: iPr; 0: tBu). The simplest magnesium complex with secondary alkyl groups is di(isopropyl)magnesium, [iPr2Mg(μ‐dx)]∞ (1′‐iPr), which has already been studied by Blasberg et al.8c Another complex with a larger dx content was accessible during crystallization in a more dioxane‐rich solution of isopropylmagnesium chloride; the molecular structure of this derivative, [{iPr2Mg(dx)}2(μ‐dx)] (1.5‐iPr), is depicted in Figure 3.
Figure 3.
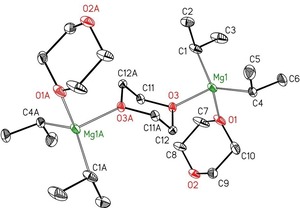
Molecular structure and numbering scheme of [{iPr2Mg(dx)}2(μ‐dx)] (1.5‐iPr). The ellipsoids represent a probability of 30 %. Hydrogen atoms have been neglected for clarity reasons. Selected bond lengths [pm]: Mg1−C1 214.0(2), Mg1−C4A 214.6(3), Mg1A−O1A 208.43(18), Mg1A−O3A 209.84(16), Mg1B−C1B 214.7(3), Mg1B−C4B 214.9(3), Mg1B−O1B 208.39(18), Mg1B−O3B 210.36(17); bond angles [°]: C1A‐Mg1A‐C4A 128.70(10), O1A‐Mg1A‐O3A 99.90(7), O3A‐Mg1A‐C1A 105.76(8), O3A‐Mg1A‐C4A 106.60(8), C1B‐Mg1B‐C4B 130.42(10), O1B‐Mg1B‐O3B 99.68(7), C1B‐Mg1B‐O3B 105.17(8), C4B‐Mg1B‐O3B 106.76(8). Atoms B not shown.
The structure is of type 1.5 and the Mg−C and Mg−O bond lengths are of the same order of magnitude as those observed for the strand structure.8c Contrary to this finding, the bond angles show significant differences. Thus, the C−Mg−C bond angles of 128.7(1) and 130.4(1)° are larger than the value of 122.19(9)° in the strand structure, and the O−Mg−O angles of 99.90(7) and 99.68(7)° are more acute in comparison with the O−Mg−O bond angle of 104.37(7)° in [iPr2Mg(μ‐dx)]∞ (1′‐iPr). Thus, for the complexes with bulky isopropyl groups, two structures, 1′ and 1.5, are observed depending on the dioxane concentration in the mother liquor, whereas for the n‐propyl congeners the coordination polymer 1′ with exclusively bridging dioxane ligands seems to be favored.
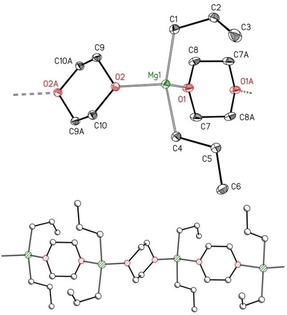
To further increase the bulkiness of the alkyl group, we studied the tert‐butylmagnesium complex. After precipitation and removal of [MgCl2(dx)2] from an ethereal solution of tert‐butylmagnesium chloride we crystallized [tBu2Mg(η1‐dx)2] (2‐tBu). The structure of this compound is depicted in Figure 4.
Figure 4.
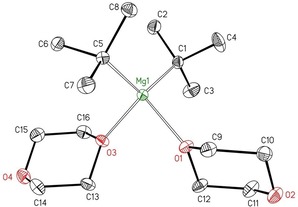
Molecular structure and numbering scheme of [tBu2Mg(η1‐dx)2] (2‐tBu). The ellipsoids represent a probability of 30 %. Hydrogen atoms have been omitted for clarity reasons. Selected bond lengths [pm]: Mg1−C1 216.72(18), Mg1−C5 217.45(18), Mg1−O1 208.71(12), Mg1−O3 207.87(8); bond angles [°]: C1‐Mg1‐C5 123.74(7), O1‐Mg1‐O3 92.04(5), O3‐Mg1‐C1 109.22(6), O1‐Mg1‐C1 107.24(6), O3‐Mg1‐C5 109.00(6), O1‐Mg1‐C5 110.98(6).
This complex is the fifth derivative of type 2. This structure type forms with sterically demanding groups and in the presence of a moderate excess of 1,4‐dioxane during crystallization. The Mg−C bond lengths of dialkylmagnesium increase in the order primary (213–215 pm) < secondary (214–216 pm) < tertiary alkyl groups (216–218 pm), whereas the C−Mg−C bond angles show no clear trend.
The crystalline 2‐tBu partially loses ligated dioxane during the drying process in vacuo. We did not isolate this dioxane‐poor congener, but structures of tert‐butylmagnesium complexes with dioxane ligands have already been observed in the heteroleptic [L2{Mg(tBu)}4(μ‐dx)2], in which L is a bidentate bridging ligand, and in the trinuclear compound [(thf)Mg(tBu)2(μ‐dx)Mg(tBu)LMg(tBu)].19
Structures of [Ar2Mg(dx)n] with increasing hindrance at the ortho‐substituted phenyl groups
The coordination polymer [Ph2Mg(μ‐dx)]∞ (1′‐Ph) is insoluble in toluene. The addition of 1,4‐dioxane to this suspension led to a clear solution. Cooling of this solution again yielded the starting [Ph2Mg(μ‐dx)]∞ (1′‐Ph). Dissolution of these crystals in pure dioxane gave another complex that is highly soluble in dx. The clear crystals of this complex turned dull after isolation and removal of the mother liquor. The molecular structure of this dioxane adduct, [Ph2Mg(dx)3] (3‐Ph), is depicted in Figure 5.
Figure 5.
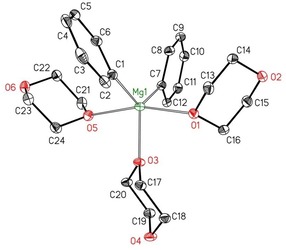
Molecular structure and numbering scheme of [Ph2Mg(dx)3] (3‐Ph). The ellipsoids represent a probability of 30 %. Hydrogen atoms have been omitted for the sake of clarity. Selected bond lengths [pm]: Mg1−C1 217.19(14), Mg1−C7 216.01(13), Mg1−O1 224.51(11), Mg1−O3 209.10(10), Mg1−O5 228.99(11); bond angles [°]: C1‐Mg1‐C7 125.08(5), O1‐Mg1‐O3 82.81(4), O1‐Mg1‐O5 164.68(5), O1‐Mg1‐C1 95.63(5), O1‐Mg1‐C7 95.87(5), O3‐Mg1‐C1 119.20(5), O3‐Mg1‐C7 115.45(5), O3‐Mg1‐O5 81.90(4), O5‐Mg1‐C1 91.07(5), O5‐Mg1‐C7 91.48(5), C2‐C1‐C6 113.35(13), C8‐C7‐C12 113.47(11).
The τ parameter, which is an indicator of the geometry of pentacoordinate metal complexes, can be calculated according to the equation τ=(β−α)/60° (in which β is the largest and α the second largest bond angle at the metal center) with τ=1 for an ideal trigonal bipyramid and τ=0 for a square pyramid.20 For 3‐Ph, a τ value of 0.66 was elucidated, which is much closer to a trigonal‐bipyramidal coordination sphere with O1 and O5 in apical positions (O1−Mg1−O5 164.68(4)°). Deviation from a linear arrangement is enforced by the steric repulsion between these dioxane ligands and the phenyl groups. The equatorial plane contains the ipso‐carbon atoms C1 and C7 and the Lewis base O3 (angle sum=359.73(5)°). The higher coordination number of the magnesium atom in 3‐Ph leads to elongated Mg−C and Mg−O bonds (Mg−C 217.19(14) and 216.01(13) pm, Mg−Oaxial 224.51(11) and 228.99(11) pm, Mg−Oequatorial 209.10(10) pm) in comparison with the strand structure [Ph2Mg(μ‐dx)]∞ (1′‐Ph; Mg−C 213.5(2) pm, Mg−O 208.1(2) and 206.1(2) pm) with tetracoordinate metal atoms.
The type 2 diphenylmagnesium complex [Ph2Mg(dx)2] is unknown even though Bickelhaupt and co‐workers were able to prepare the corresponding THF adduct [Ph2Mg(thf)2].2a
Ortho substitution with methyl groups increases the steric requirements of the aryl groups and a coordination number of 5 could not be achieved. Consequently, addition of dioxane to an ethereal solution of o‐tolylmagnesium bromide and removal of [MgBr2(dx)2] allowed the crystallization and isolation of [(oTol)2Mg(dx)2] (2‐oTol) but not a strand‐like structure of type 1′ as found for diphenylmagnesium. The molecular structure of 2‐oTol is depicted in Figure 6.
Figure 6.
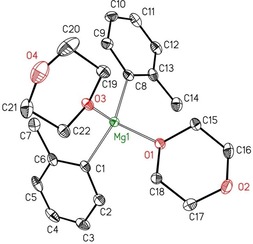
Molecular structure and numbering scheme of [(oTol)2Mg(dx)2] (2‐oTol). The ellipsoids represent a probability of 30 %. Hydrogen atoms have been neglected for reasons of clarity. Selected bond lengths [pm]: Mg1−C1 215.6(2), Mg1−C8 214.9(2), Mg1−O1 200.00(16), Mg1−O3 205.88(17); bond angles [°]: C1‐Mg1‐C8 124.39(9), O1‐Mg1‐O3 92.60(7), O1‐Mg1‐C1 107.01(9), O3‐Mg1‐C1 109.13(8), O2‐Mg1‐C8 111.49(8), O3‐Mg1‐C8 107.58(9), C2‐C1‐C6 115.1(2), C9‐C8‐C13 114.7(2).
Whereas the magnesium atom in the phenyl derivative 3‐Ph has a coordination number of 5, the bulkier oTol group only allows tetracoordinate magnesium centers. The Mg−C bond lengths in the distorted tetrahedral complexes of type 2 increase with increasing steric pressure in the order phenyl (213.5(2) pm)<o‐tolyl (214.9(2)–215.6(2) pm)<2,4,6‐trimethylphenyl (mesityl, 216.7(2) pm).7 The C−Mg−C bond angles vary from 117.55(9)° (Mes) to 123.96(8)° (Ph) to 124.39(9)° (oTol).
The complex [(oTol)2Mg(dx)2] (2‐oTol) is soluble in warm toluene. From this solution, tetracoordinate type 1.5 magnesium centers were also accessible. The molecular structure of [{(oTol)2Mg(dx)}2(μ‐dx)] (1.5‐oTol) is shown in Figure 7. The o‐methyl substituents are disordered but crystallization as a type 1.5 complex was verified unequivocally. The two magnesium atoms are in distorted tetrahedral environments with a bridging dx ligand.
Figure 7.
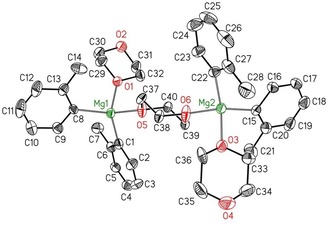
Molecular structure and numbering scheme of [{(oTol)2Mg(dx)}2(μ‐dx)] (1.5‐oTol). The ellipsoids represent a probability of 30 %. Hydrogen atoms and the disorder of the tolyl groups and dioxane ligand have been omitted for the sake of clarity. Selected bond lengths [pm]: Mg1−C1 214.0(2), Mg1−C8 217.6(4), Mg1−O1 207.4(3), Mg1−O5 209.7(3), Mg2−C15 213.2(4), Mg2−C22 216.7(3), Mg2−O3 219.2(19), Mg2−O6 207.2(3); bond angles [°]: C1‐Mg1‐C8 107.22(18), C15‐Mg2‐C22 118.65(17), O1‐Mg1‐O5 95.41(13), O3‐Mg2‐O6 101.3(5), C6‐C1‐C2 113.3(4), C16‐C15‐C20 114.0(4).
Dimesitylmagnesium (bis(2,4,6‐trimethylphenyl)magnesium) crystallized as [Mes2Mg(dx)2] (2‐Mes) from an ethereal solution of mesitylmagnesium bromide after addition of 1,4‐dioxane and removal of magnesium bromide.7 Furthermore, the structure of 2‐Mes is comparable to the structures of the thf adducts of dimesityl‐ ([Mes2Mg(thf)2]) and bis(2,4,6‐triisopropylphenyl)magnesium ([Trip2Mg(thf)2]).21 Contrary to this finding, bis[2,4,6‐tri(tert‐butyl)phenyl]magnesium forms no stable adducts with tetrahydrofuran.15b
1,4‐Dioxane adducts of coordination polymers with layer structure
After the investigation of di(hydrocarbyl)magnesium complexes with sp3‐ (alkyl) and sp2‐hybridized (aryl) carbon atoms, we also studied congeners with an sp‐hybridized carbon atom. Compounds of the type [(R‐C≡C)2Mg(dx)n] (n=1.5 (SiMe3) and 2 (Ph)) are nearly insoluble in diethyl ether, dioxane, and toluene, but soluble in THF. From such a solution, [(Ph‐C≡C)2Mg(thf)4] was isolated with a distorted octahedral environment of the magnesium center.22
For solubility reasons we prepared an alkynyl complex by the deprotonation of trimethylsilylacetylene with [nPr2Mg(μ‐dx)]∞ (1′‐nPr) in excess dioxane to yield a compound with the composition [(Me3Si‐C≡C)2Mg(dx)1.5]. This complex is soluble in THF but only very sparingly soluble in dioxane and insoluble in hydrocarbons, which is quite unique for a complex of type 1.5. The 13C{1H} NMR spectrum shows two characteristic resonances at δ=112.1 and 158.5 ppm for the ethynyl fragment. To grow single crystals of this complex we stored a mixture of [nBu2Mg(μ‐dx)] (1‐nBu) and trimethylsilylacetylene in 1,4‐dioxane at 5 °C. This procedure allowed the slow crystallization of [(Me3Si‐C≡C)2Mg(dx)1.5]∞ (1.5′‐C2SiMe3), but the crystal quality was very poor. Therefore, we could only elucidate a structural motif. The structure is depicted in Figure 8.
Figure 8.
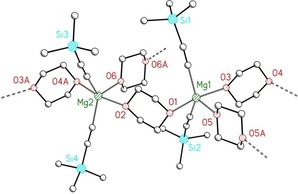
Structural motif of [(Me3Si‐C≡C)2Mg(dx)1.5]∞ (1.5′‐C2SiMe3). The atoms are shown with arbitrary radii and hydrogen atoms have been neglected for reasons of clarity.
Despite poor crystal quality and a substandard data set, the structural motif was deduced unequivocally and revealed the solid‐state structure. The pentacoordinate magnesium atoms are embedded in trigonal‐bipyramidal environments with the alkynyl groups in apical positions. The 1,4‐dioxane ligands with chair conformations occupy equatorial positions and act as bridging ligands between magnesium atoms, leading to a layer structure. As a consequence of this packing, the magnesium atoms form a layer structure of regular six‐membered rings. The alkynyl ligands are bound above and below the layer and establish borders between two layers. Okuda and co‐workers23 determined the structure of [(all)2Mg(thf)(μ‐dx)]∞ (all=allyl); the slim allyl groups here would have also allowed the formation of a layer structure, but two‐dimensional (2D) binding is prevented by the presence of the thf ligands instead of dx molecules. Therefore, a strand structure is observed in the crystalline state. A 2D network was found for the heterobimetallic compound [{M(all)3Mg(all)2}2(μ‐dx)5]∞ with M=Y and La.24 In [(Me3Si‐C≡C)2Mg(thf)4], all the dx ligands are substituted by thf ligands leading to the breakup of the coordination polymer to yield discrete molecular complexes with hexacoordinate magnesium atoms.25
The metalation of phenylacetylene with bis(dioxane)di(tert‐butyl)magnesium (2‐tBu) in diethyl ether yielded quantitatively [(Ph‐C≡C)2Mg(dx)2]∞ (2′‐C2Ph), which is nearly insoluble in 1,4‐dioxane. The NMR spectra reveal that two dioxane molecules are bound to the magnesium center. The crystal structure of [(Ph‐C≡C)2Mg(dx)2]∞ (2′‐C2Ph) verifies that the magnesium atoms are embedded in octahedral environments forming a 2D network. The structure is depicted in Figure 9. The phenylethynyl groups are bound above and below the layer formed by the magnesium atoms and the bridging ether molecules.
Figure 9.
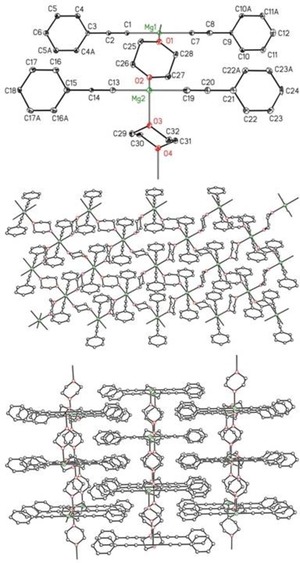
Molecular structure (top) and cutout of the layer structure of [(Ph‐C≡C)2Mg(dx)2]∞ (2′‐C2Ph, middle). The ellipsoids represent a probability of 30 %. Hydrogen atoms have been omitted for the sake of clarity. The stacking of the layers is depicted at the bottom.
Finally, we also investigated the 1,4‐dioxane adducts of magnesium chloride and bromide, which precipitated during the addition of dioxane to solutions of Grignard reagents. Extremely sparingly soluble [MgBr2(dx)2]∞ (2′‐Br) was tempered in warm tetrahydrofuran leading to a single crystalline solid. This observation is in agreement with the expectation that THF and dx exhibit comparable basicity leading to very low concentrations of soluble [MgBr2(thf)4], which has been characterized previously.26, 27 This procedure allowed us to grow single crystals of sufficient quality for X‐ray crystal‐structure determination. Homologous [MgCl2(dx)2]∞ (2′‐Cl) exhibited comparable crystallization properties.
The molecular structure of the coordination polymer [MgBr2(dx)2]∞ (2′‐Br) with a layer structure is depicted in Figure 10; the analogous structure of [MgCl2(dx)2]∞ (2′‐Cl) is presented in the Supporting Information. The magnesium centers in 2′‐Br are embedded in octahedral environments with trans‐arranged halide ions. The environments of the magnesium centers are very similar to that in the molecular tetrahydrofuran adduct [MgBr2(thf)4] with average Mg−O and Mg−Br bond lengths of 216 and 262.5 pm, respectively.26, 27 In the coordination polymer [Mg(μ‐Br)2(thf)2]∞, which has been crystallized from a mixture of dichloromethane and hexane, the bromine atoms occupy bridging positions leading to a coordination polymer with rather similar Mg−O and Mg−Br distances of 212.6 and 263.3 pm, respectively.27
Figure 10.
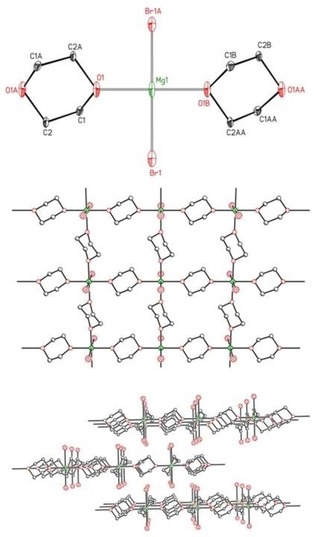
Molecular structure (top) and cutout of the layer structure of [MgBr2(dx)2]∞ (2′‐Br, middle). The ellipsoids represent a probability of 30 %. Hydrogen atoms have been omitted for the sake of clarity. The stacking of the layers is shown at the bottom.
The Mg(dx) networks of the layer structures of [(Me3Si‐C≡C)2Mg(dx)1.5]∞ (1.5′‐C2SiMe3) and [MgBr2(dx)2]∞ (2′‐Br) are compared in Figure 11. The groups R are positioned above and below the magnesium atoms, leading to penta‐ and hexacoordinated metal centers, respectively. The steric requirements of the R groups in 1.5′‐C2SiMe3 lead to a honeycomb structure, whereas the smaller bromine atoms in 2′‐Br lead to a tessellated structure. The directing influence exerted by the steric demand of the groups R is evident in Figure 11, in which the same Mg⋅⋅⋅Mg distances are indicated by the solid lines (symbolizing the bridging dx ligands).
Figure 11.
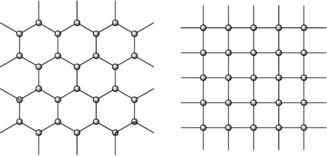
Structural diversity of the coordination polymers [(Me3Si‐C≡C)2Mg(dx)1.5]∞ (1.5′‐C2SiMe3, left) and [MgR2(dx)2]∞ (2′‐C2Ph, 2′‐Cl, and 2′‐Br, right) with penta‐ and hexacoordinate metal centers, respectively. The circles at the knots of the network represent the magnesium atoms and the connecting lines symbolize the bridging 1,4‐dioxane ligands.
Substitution of dx in [MgBr2(dx)2]∞ (2′‐Br)
Due to the fact that [MgBr2(dx)2]∞ (2′‐Br) is nearly insoluble in common organic solvents, several attempts were undertaken to recrystallize this coordination polymer. Crystallization of 2′‐Br from a solvent mixture of DMF and dioxane led to the precipitation of [{(dmf)6Mg}Br2] (A). The molecular structure contains a centrosymmetric cation with a hexacoordinate metal center and clearly separated bromide ions (Mg⋅⋅⋅Br 527.7(3) and 678.2(3) pm, see the Supporting Information). Traces of water also led to the precipitation of a few crystals of the more soluble [{trans‐(H2O)2Mg(dmf)4}Br2]∞ (B, see the Supporting Information). In this latter centrosymmetric molecule, the magnesium atom is in an octahedral environment, coordinated to the oxygen donors of the dmf ligands (Mg−O 202.86(10) and 208.26(10) pm) and water (Mg−O 209.89(11) pm). The bromide ions are again separated from the cations (Mg⋅⋅⋅Br 480.8 and 493.8 pm), but are bound at the periphery through hydrogen bridges to the water ligands (Br⋅⋅⋅H 248.6 and 252.1 pm). This mode of coordination leads to the formation of a coordination polymer with a strand structure. Extraction of [MgBr2(dx)2]∞ (2′‐Br) with hot 1,2‐dimethoxyethane (dme) only allowed the isolation of the already known [cis‐Br2Mg(dme)2] (C).28 Tridentate diglyme also forms very stable molecular complexes with MgBr2, namely [(diglyme)Mg(Br)(μ‐Br)]2 (D) and [(diglyme)(thf)MgBr2] (E) with distorted octahedrally coordinated metal centers.29
Conclusions
In this work we have systematically studied the synthesis of di(hydrocarbyl)magnesium complexes from ethereal solutions of Grignard reagents by the addition of 1,4‐dioxane. With very few exceptions, in which the Schlenk equilibrium lies completely on the side of heteroleptic RMgX complexes (such as [(Me3Si)2C{MgBr(dx)2}2]),6 the dioxane method provides advantageous access to soluble R2M compounds. The type of crystalline [R2Mg(dx)n] adduct depends on the molar ratio of R2Mg/dx in solution and on the hydrocarbyl group R, which influences the solubility and steric requirements. The structure types of the isolated crystalline derivatives are summarized in Table 1.
Table 1.
Structure types of crystalline [R2Mg(dx)n] with coordination numbers of the MgII atoms (C.N.(Mg)) of 4 (blue), 5 (red), and 6 (green) depending on the alkyl and aryl substituents R.
|
|
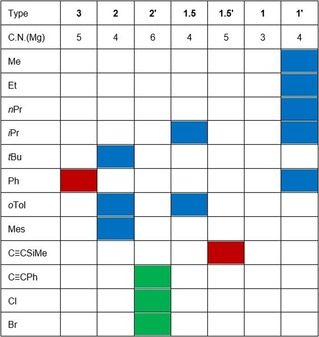
High dioxane concentrations and bulkier organic groups promote the formation of highly soluble mononuclear compounds of structure types 3 (R=Ph) and 2 (R=tBu, oTol, and Mes7). In some cases with substituents of intermediate size (R=iPr, Bz,7 and oTol) dinuclear complexes of type 1.5 are accessible. This structure type can aggregate to form coordination polymers of type 1′ if the organic groups exhibit a medium‐to‐small size and if the concentration of 1,4‐dioxane during crystallization is rather low. For selected medium‐sized derivatives (R=iPr, Ph, and oTol) it was possible to isolate two different structures in the solid state. R2Mg congeners with extremely bulky groups (R=C(SiMe3)3 and Mes*)15 crystallize without co‐ligands from dioxane‐containing solutions. A structurally authenticated mono‐dioxane adduct of a di(hydrocarbyl)magnesium complex of type 1 with a three‐coordinate metal center is as yet unknown.
The exchange equilibria with 1,4‐dioxane, which depend on complex concentration, MgR2/dioxane ratio, and the nature of the group R, are depicted in Scheme 5. Only those derivatives that exceed the saturation concentration and crystallize are accessible by this method. Therefore, we cannot exclude a richer coordination chemistry of di(hydrocarbyl)magnesium–dioxane adducts with other nuclearities. The complexes R2Mg without co‐ligands can be stabilized with very bulky groups R or isolated after removal of dioxane in vacuo. The latter procedure even allowed the isolation of dimethylmagnesium as a coordination polymer with tetracoordinate magnesium atoms.16
Scheme 5.

Structural diversity of the hitherto known 1,4‐dioxane adducts of diorganylmagnesium complexes authenticated by X‐ray crystal‐structure determinations (complexes with tetracoordinate Mg are shown in blue, with pentacoordinate Mg in red, and with hexacoordinate Mg in green). The left column shows molecular complexes and solvation/desolvation reactions, the right column contains the coordination polymers arising from aggregation and deaggregation equilibria.
The aggregation of mononuclear [R2Mg(dx)2] would lead to the formation of a coordination polymer with hexacoordinate magnesium atoms. thf adducts with the composition [R2Mg(thf)4] are known that require very slim ligands to allow an octahedral environment. The substitution of two thf ligands by a bridging dioxane molecule enables the formation of coordination polymers with hexacoordinate magnesium centers.
The solubility of the magnesium complexes is strongly related to their aggregation chemistry. Molecular complexes of the types 1.5‐R, 2‐R, and 3‐R are highly soluble in ethereal solvents. The formation of strand structures reduces the solubility and compounds of type 1′‐R are sparingly soluble. Complexes of the types 1.5′‐R and 2′‐R form layer structures and are insoluble in common organic solvents. The degree of aggregation is influenced by the size and steric requirements of R, with small R ligands leading to higher coordination numbers of magnesium and higher degrees of aggregation.
Experimental Section
General
All manipulations were carried out under anaerobic conditions in an argon atmosphere using standard Schlenk techniques. The solvents were dried according to common procedures and distilled in an argon atmosphere; deuterated solvents were dried over sodium, degassed, and saturated with argon. The yields given are not optimized. The magnesium contents of the compounds were determined by complexometric titrations with Eriochrom Black T.30 The alkalinities of the solid diorganylmagnesium compounds were determined after hydrolysis of a specific amount in ice/water by acidimetric titration with 0.1 n H2SO4 against phenolphthalein. 1H, 13C{1H}, and 29Si{1H} NMR spectra were recorded on a Bruker AC 400 spectrometer at given temperatures. Chemical shifts are reported in parts per million (ppm, δ scale) relative to the residual signal of the solvent.31 Due to fast [D8]THF/dx exchange reactions in solution, only resonances of noncoordinated dioxane were detected in the NMR spectra.
Trimethylsilylacetylene (98 %) and tert‐butyl chloride (95–98 %) were supplied by ABCR GmbH, 1.0 m phenylmagnesium bromide solution in THF by Merck, o‐bromotoluene (98 %) by Lancaster, isopropyl chloride by Fluka, n‐butyl chloride by Riedel de Haën, n‐propyl bromide by Acros, and 3.0 m methylmagnesium chloride solution in THF by Sigma–Aldrich.
Synthesis and characterization
Synthesis of [(CH3)2Mg(μ‐dx)]∞ (1′‐Me): A solution of commercial 3.0 m methylmagnesium chloride in THF (17 mL, 51 mmol) was diluted with THF (50 mL). The slow addition of 1,4‐dioxane (18 mL, 204 mmol) led to the precipitation of [MgCl2(dx)2]. The reaction mixture was stored overnight at room temperature and then filtered through a frit covered with diatomaceous earth. A yield of 87 % was determined by titration of an aliquot of the filtrate with 0.1 n HCl. All volatiles were removed in vacuo and the residue evacuated until dryness. The residue was dissolved in dioxane (30 mL) and this suspension was heated at reflux for 10 min. Then, the hot solution was filtered through a Schlenk frit covered with diatomaceous earth. During cooling of the filtrate colorless crystals precipitated. These crystals were collected on a frit, washed with diethyl ether, and dried in vacuo. Yield: 1.3 g of 1′‐Me (35.8 % relative to CH3MgCl). During hydrolysis of this compound with ice/water, inflammation occurred. Therefore, a specific amount of this compound was dissolved in THF and this solution was carefully hydrolyzed to quantitatively determine the metal content. Alkalinity: calcd: 688.3 mg H2SO4 g−1; found: 691.8 mg; 1H NMR (400.1 MHz, [D8]THF): δ=−1.82 (s, 6 H; Mg‐CH3), 3.54 ppm (s, 8 H; dx); 13C NMR (100.6 MHz, [D8]THF): δ=−16.5 (q, 1 J CH=105.6 Hz, Mg‐CH3), 67.9 ppm (t, 1 J CH=142.3 Hz, O‐CH2, dx); elemental analysis calcd (%) for C6H14MgO2 (142.5): Mg 17.06; found: Mg 16.98.
Synthesis of [(n‐C3H7)2Mg(μ‐dx)]∞ (1′‐nPr): A solution of n‐propylmagnesium bromide was prepared from magnesium turnings (3.0 g, 123.4 mmol) and n‐propyl bromide (12.4 g, 100.8 mmol) in diethyl ether (100 mL; yield: 82 %). 1,4‐Dioxane (16 mL, 182 mmol) was then added dropwise to this reaction mixture. During this highly exothermic procedure a colorless precipitate formed. After resting overnight the precipitate was removed by means of a Schlenk frit covered with diatomaceous earth. An aliquot of the filtrate was titrated with 0.1 m HCl (yield: 64 %). The volume of the solution was reduced to a fifth of the original volume and stored in a refrigerator at −40 °C. The colorless crystals of 1′‐nPr precipitated from this mother liquor were washed with very cold diethyl ether and dried in vacuo. Yield: 1.1 g of 1′‐nPr (11.0 % relative to n‐propyl bromide); alkalinity: calcd: 493.9 mg H2SO4 g−1; found: 486.5 mg; 1H NMR (400.1 MHz, [D8]THF): δ=−0.64 (t, J=7.8 Hz, 4 H; CH2Mg), 0.87 (t, J=7.2 Hz, 6 H; CH3), 1.54 (m, 4 H; CH2), 3.55 ppm (s, 8 H; dx); 13C{1H} NMR (100.6 MHz, [D8]THF): δ=12.2 (CH2Mg), 23.7 (CH3), 24.3 (CH2), 67.8 ppm (dx); elemental analysis calcd (%) for C10H22MgO2 (198.6): Mg 12.24; found: Mg 12.07.
Synthesis of [(n‐C4H9)2Mg(dx)] (1‐nBu): A solution of n‐butylmagnesium chloride was prepared in diethyl ether (200 mL) from magnesium turnings (6.0 g, 0.247 mol) and n‐butyl chloride (19.5 g, 0.211 mol; yield: 82 %). 1,4‐Dioxane (22 mL, 0.25 mol) was then added dropwise to this solution. During this highly exothermic procedure a colorless solid precipitated. After standing overnight the precipitate was removed by means of a Schlenk frit covered with diatomaceous earth. An aliquot of the filtrate was titrated with 0.1 m HCl (yield: 59 %). All volatiles were removed and the residue dried in vacuo. Then diethyl ether (10 mL) was added and this solution was stored at −78 °C. A colorless amorphous precipitate formed that was collected and dried in vacuo. Yield: 2.4 g of 1‐nBu (10.3 % relative to n‐butyl chloride); alkalinity: calcd: 432.7 mg H2SO4 g−1; found: 422.9 mg; 1H NMR (400.1 MHz, [D8]THF): δ=−0.70 (t, J=8.0 Hz, 4 H; CH2Mg), 0.80 (t, J=7.3 Hz, 6 H; CH3), 1.19 (m, 4 H; γ‐CH2), 1.47 (m, 4 H; β‐CH2), 3.54 ppm (s, 8 H; dx); 13C{1H} NMR (100.6 MHz, [D8]THF): δ=8.2 (CH2Mg), 14.7 (CH3), 32.4 (CH2), 34.0 (CH2), 67.8 ppm (dx); elemental analysis calcd (%) for C12H26MgO2 (226.6): Mg 10.73; found: Mg 10.49.
Synthesis of [{(iPr)2Mg(η1‐dx)}2 ( μ‐dx)] (1.5‐iPr): A 1.5 m isopropylmagnesium chloride solution was prepared from magnesium turnings (6.0 g, 0.247 mol) and isopropyl chloride (24.0 g, 0.195 mol) in diethyl ether (100 mL; yield: 55 %). 1,4‐Dioxane (28 mL, 0.318 mol) was then slowly added at room temperature. During this procedure a colorless solid precipitated. This suspension was stored overnight. Then, the precipitate was removed by means of a Schlenk frit covered with diatomaceous earth and washed with diethyl ether. The filtrate contained the product (yield: 34 %). At −20 °C, [{(iPr)2Mg(dx)}2(μ‐dx)] precipitated as colorless, air‐ and moisture‐sensitive crystals. Yield: 7.8 g of 1.5‐iPr (33 % relative to isopropyl chloride); alkalinity: calcd: 404.2 mg H2SO4 g−1; found: 396.9 mg. 1H NMR (400.1 MHz, [D8]THF): δ=−0.40 (sept, J=7.8 Hz, 4 H; Mg‐CH), 1.28 (d, J=7.8 Hz, 24 H; CH3), 3.61 ppm (s, 24 H; dx); 13C{1H} NMR (100.6 MHz, [D8]THF): δ=10.0 (Mg‐CH), 26.3 (CH3), 67.8 ppm (dx); elemental analysis calcd (%) for C24H52Mg2O6 (485.3): Mg 10.02; found: Mg 9.83.
Synthesis of [{(oTol)2Mg(η1‐dx)}2(μ‐dx)] (1.5‐oTol): 2‐oTol (1.8 g, 4.70 mol; see below for the preparation of 2‐oTol) was suspended in toluene (10 mL) and the mixture stirred and warmed until a clear solution formed. Then, the stirring was finished and the solution kept in a warm water bath. Colorless crystals of the product precipitated overnight. The crystals were collected on a Schlenk frit, washed with diethyl ether, and dried in vacuo. Yield: 1.40 g of colorless crystals of 1.5‐oTol (88 % with respect to the initially used 2‐oTol). The compound is highly soluble in THF but sparingly soluble in toluene and diethyl ether. Alkalinity: calcd: 289.5 mg H2SO4 g−1; found: 278.9 mg; 1H NMR (400.1 MHz, [D8]THF): δ=2.38 (s, 12 H; CH3), 3.54 (s, 24 H; dx), 6.78–6.79 (t, 4 H; 4‐H), 6.80–6.832 (td, 4 H; 5‐H), 6.84–6.90 (dd, 4 H; 3‐H), 7.53–7.54 ppm (dd, 4 H; 6‐H); 13C{1H} NMR (150.9 MHz, [D8]THF): δ=28.4 (CH3), 67.8 (dx), 123.3 (C‐4), 124.9 (C‐5), 126.5 (C‐3), 140.7 (C‐6), 147.8 (C‐2‐C), 169.2 ppm (C‐1); elemental analysis calcd (%) for C40H52Mg2O6 (677.4): calcd: Mg 7.18; found: Mg 7.09.
Synthesis of [(tBu)2Mg(η1‐dx)2] (2‐tBu): 1,4‐Dioxane (30 mL, 0.341 mol) was slowly added to a 1.08 m tert‐butylmagnesium chloride solution (100 mL) in diethyl ether. During this procedure a colorless solid precipitated. The suspension was stored overnight at room temperature Then, the precipitate was removed by means of a Schlenk frit covered with diatomaceous earth and washed with diethyl ether. Acidimetric titration of an aliquot gave a yield of 33 %. The filtrate was concentrated till crystallization started and then stored in a refrigerator. This suspension was mixed with heptane (20 mL) and the precipitate collected on a frit and washed with heptane. The solid was briefly dried in vacuo because it lost ligated dioxane when exposed to a vacuum. Yield: 9.0 g of colorless, air‐ and moisture‐sensitive crystals of 2‐tBu (21.5 % relative to the initially used tert‐butyl chloride); alkalinity: calcd: 311.6 mg H2SO4 g−1; found: 318.0 mg; 1H NMR (400.1 MHz, [D8]THF): δ=0.86 (s, 18 H; CH3), 3.54 ppm (s, 10 H; dx); 13C{1H} NMR (100.6 MHz, [D8]THF): δ=15.7 (C‐Mg), 35.8 (CH3), 67.8 ppm (dx); elemental analysis calcd (%) for C16H34MgO4 (314.7): Mg 7.72; found: 7.88. Single crystals were grown from a solvent mixture of diethyl ether and 1,4‐dioxane.
Synthesis of [(oTol)2Mg(η1‐dx)2] (2‐oTol): Magnesium turnings (3.0 g, 123 mmol) were suspended in diethyl ether (100 mL). Thereafter, o‐tolyl bromide (17.09 g, 99 mmol) was slowly added to the mixture in portions of 2 mL. The reaction was initiated by the addition of some drops of 1,2‐dibromoethane. After the reaction seemed to cease, the mixture was heated at reflux for 1 h. Then, the solution was filtered through a Schlenk frit (filled with anhydrous diatomaceous earth; yield: 67 %, as calculated from titration of an aliquot with 0.1 n H2SO4). The filtrate was cooled to room temperature and then dioxane (13 mL, 122 mmol) was slowly added in 2 mL portions. The reaction mixture was kept overnight at room temperature and precipitated [MgBr2(dx)2] was removed by filtration through a Schlenk frit (titrated yield: 17.4 %). Pure 2‐oTol crystallized from the filtrate at 5 °C. Yield: 2.91 g of colorless crystals of 2‐o‐Tol (15.2 % with respect to the initially used o‐tolyl bromide); alkalinity: calcd: 256.2 mg H2SO4 g−1; found: 250.8 mg; 1H NMR (400.1 MHz, [D8]THF): δ=2.38 (s, 6 H; CH3), 3.54 (s, 16 H; dx), 6.78–6.79 (t, 2 H; 4‐H), 6.80–6.832 (td, 2 H; 5‐H), 6.84–6.90 (dd, 2 H; 3‐H), 7.53–7.54 ppm (dd, 2 H; 6‐H); 13C{1H} NMR (150.9 MHz, [D8]THF): δ=28.4 (CH3), 67.8 (dx), 123.3 (C‐4), 124.9 (C‐5), 126.5 (C‐3), 140.7 (C‐6), 147.8 (C‐2), 169.2 ppm (C‐1); elemental analysis calcd (%) for C22H30MgO4 (382.8): Mg 6.35; found: Mg 6.22.
Synthesis of [Ph2Mg(η1‐dx)3] (3‐Ph): A commercially available 1.0 m PhMgBr solution in THF (50 mL, 50 mmol) and dioxane (50 mL) were combined and the mixture heated at reflux. During this procedure a colorless solid precipitated, which was removed by means of a Schlenk frit. All volatiles of the filtrate were removed in vacuo. The dry residue was dissolved in boiling dioxane (50 mL) and filtered through a Schlenk frit covered with diatomaceous earth. Then, toluene (5 mL) was added to the filtrate and the volume reduced until crystallization started. At room temperature the majority of the colorless compound crystallized. These crystals were collected on a Schlenk frit, washed with cold diethyl ether, and briefly dried in vacuo. Yield: 2.1 g of 3‐Ph (19 % relative to initially used phenylmagnesium bromide solution); alkalinity: calcd: 221.5 mg H2SO4 g−1; found: 224.9 mg; 1H NMR (400.1 MHz, [D8]THF): δ=3.55 (s, 24 H; dx), 6.86–6.90 (m, 2 H; p‐H), 6.94–6.99 (m, 4 H; m‐H), 7.67–7.70 ppm (m, 4 H; o‐H); 13C{1H} NMR (100.6 MHz, [D8]THF): δ=67.8 (dx), 124.4 (C‐p), 126.3 (C‐m), 141.3 (C‐o), 170.3 ppm (C‐i); elemental analysis calcd (%) for C24H34MgO6 (442.8): Mg 5.49; found: Mg 5.57.
Synthesis of [(Me3Si‐C≡C)2Mg(μ‐dx)1.5]∞ (1.5′‐C2SiMe3): Trimethylsilylacetylene (4.3 g, 43.78 mmol) and dioxane (4.0 mL, 45.45 mmol) were added at room temperature to a 0.87 m solution of 1′‐nPr in diethyl ether (50 mL, 43.5 mmol). Shortly thereafter, a microcrystalline solid of 1.5′‐C2SiMe3 formed. This precipitate was collected on a Schlenk frit, thoroughly washed with diethyl ether, and dried in vacuum. Yield: 5.30 g of 1.5′‐C2SiMe3 (69 % relative to the initially used trimethylsilylacetylene); alkalinity: calcd: 279.5 mg of H2SO4 g−1; found: 287.8 mg. The crystals slowly lost dioxane at room temperature once isolated and therefore slowly became dull leading to enhanced magnesium values and alkalinities. 1H NMR (400.1 MHz, [D8]THF): δ=−0.04 (s, 18 H; SiMe3; 29Si satellites 2 J HSi=6.5 Hz), 3.56 ppm (s, 12 H; dx); 13C{1H} NMR (100.6 MHz, [D8]THF): δ=1.8 (SiMe3), 67.8 (dx), 112.1 (≡C‐Si), 158.4 ppm (≡C‐Mg); 29Si{1H} NMR (79.5 MHz, [D8]THF): δ=−29.2 ppm; elemental analysis calcd (%) for C32H60Mg2O6Si4 (701.7): Mg 6.92; found: Mg 7.11. Single crystals of 1.5′‐C2SiMe3 were obtained when trimethylsilylacetylene (1.2 g, 12.2 mmol) and dioxane (1.0 mL, 11.4 mmol) were added to a 0.36 m solution of [(n‐C4H9)2Mg(dx)] (12.6 mmol) in diethyl ether (35 mL) at −20 °C and stored overnight in a refrigerator at 5 °C.
Synthesis of [(Ph‐C≡C)2Mg(dx)2]∞ (2′‐C2Ph): A freshly prepared 0.365 m solution of [(nBu)2Mg(dx)] in diethyl ether (90 mL; see above) was cooled to 0 °C. Phenylacetylene (6.4 g, 62.7 mmol) was added dropwise to this solution with stirring. Evolution of a colorless gas was observed as the drops of phenylacetylene hit the solution. After the addition of half of the Ph‐C≡C‐H a colorless precipitate formed. After complete addition of phenylacetylene the reaction mixture was stirred at room temperature for an additional hour. The precipitate was collected on a Schlenk frit, washed with diethyl ether, and dried in vacuo. Yield: 12.13 g (96 % relative to the initially used phenylacetylene) of 2′‐C2Ph; alkalinity: calcd: 243.3 mg H2SO4 g−1; found: 243.5 mg; 1H NMR (400.1 MHz, [D8]THF): δ=3.58 (s, 16 H; dx), 6.91 (m, 2 H; p‐H), 7.08 (m, 4 H; m‐H), 7.20 ppm (m, 4 H; o‐H); 13C{1H} NMR (100.6 MHz, [D8]THF): δ=67.9 (dx), 109.2 (≡C‐), 124.9 (C‐p), 128.2 (C‐m), 130.6 (C‐i), 131.3 (C‐o), 131.8 ppm (≡C‐Mg); elemental analysis calcd (%) for C24H26MgO4 (402.7): Mg 5.96; found: Mg 6.03. Single crystals of 2′‐C2Ph were grown by heating a suspension of the microcrystalline substance (0.66 g, 1.6 mmol) in a mixture of THF (10 mL) and dioxane (4.5 mL, 51.1 mmol) at 65 °C for 72 h.
Crystallography
Crystallization of [MgCl2(μ‐dx)2]∞ (2′‐Cl): A 3.0 m solution of methylmagnesium chloride (3.0 mL, 9.0 mmol) in THF was diluted with THF (25 mL). 1,4‐Dioxane (2 mL, 22.7 mmol) was added to this solution. The clear solution was then heated at 65 °C in a 270 mL Schlenk tube to yield a white amorphous precipitate after a few minutes. After 1 week at this temperature, the precipitate transformed into colorless crystals of 2′‐Cl of different size.
Crystallization of [MgBr2(μ‐dx)2]∞ (2′‐Br): A 0.9 m solution of n‐propylmagnesium bromide (3.0 mL, 2.7 mmol) in THF was diluted with THF (25 mL). Then, 1,4‐dioxane (0.9 mL, 10.2 mmol) was added. The clear solution was heated at 65 °C. During this procedure a colorless precipitate of 2′‐Br slowly formed. This suspension was tempered for 1 week at this temperature. During this time, the precipitate aged and turned into a crystalline solid with crystals of different size.
Crystal structure determinations: The intensity data for the compounds were collected on a Nonius KappaCCD diffractometer using graphite‐monochromated MoKα irradiation. The data were corrected for Lorentzian and polarization effects; absorption was taken into account on a semi‐empirical basis using multiple scans.32, 33, 34 The structures were solved by direct methods (SHELXS)35 and refined by full‐matrix least‐squares techniques against F o 2 (SHELXL‐97).36 The hydrogen atoms of the compounds 1′‐Me, 1′‐nPr, 2‐tBu, and B were located by difference Fourier synthesis and refined isotropically. All other hydrogen atoms were included at calculated positions with fixed thermal parameters. The crystal of 2′‐C2Ph was a non‐merohedral twin. The twin law was determined by PLATON37 as (0.003 0.997 0.000) (1.003 −0.003 0.000) (0.000 0.000 −1.000). The contribution of the main component was refined to 0.845(1). All non‐disordered, non‐hydrogen atoms were refined anisotropically.36 The crystals of 1.5′‐C2SiMe3 were extremely thin and/or of low quality, resulting in a substandard data set; however, the structure is of sufficient quality to show connectivity and geometry despite the high final R value. We only publish here the conformation of the molecule and the crystallographic data. We will not deposit the data at the Cambridge Crystallographic Data Centre. The crystallographic data as well as structure solution and refinement details are summarized in Table S1 in the Supporting Information). XP38 and POV‐Ray39 software were used for structure representations.
CCDC https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/chem.201903120 contain the supplementary crystallographic data for this paper. These data are provided free of charge by http://www.ccdc.cam.ac.uk/.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
P.R.M. is the recipient of a generous stipend of the Studienstiftung des deutschen Volkes (Bonn, Germany, grant no 366961). We acknowledge the valuable support of the NMR service platform (http://www.nmr.uni-jena.de/) of the Faculty of Chemistry and Earth Sciences of the Friedrich Schiller University Jena, Germany.
R. Fischer, H. Görls, P. R. Meisinger, R. Suxdorf, M. Westerhausen, Chem. Eur. J. 2019, 25, 12830.
Contributor Information
Dr. Reinald Fischer, http://www.lsac1.uni-jena.de
Prof. Dr. Matthias Westerhausen, Email: m.we@uni-jena.de.
References
- 1.
- 1a. Cope A. C., J. Am. Chem. Soc. 1935, 57, 2238–2239; [Google Scholar]
- 1b. Wakefield: B. J. Organomagnesium Methods in Organic Synthesis Academic Press, London, 1995; [Google Scholar]
- 1c. Handbook of Grignard Reagents (Eds.: G. S. Silverman, P. E. Rakita), CRC, New York, 1996; [Google Scholar]
- 1d. Grignard Reagents: New Developments (Ed.: H. G. Richey), Wiley, Chichester, 2000; [Google Scholar]
- 1e. The Chemistry of Organomagnesium Compounds (Patai Series: The Chemistry of Functional Groups) (Eds.: Z. Rappoport, I. Marek), Wiley, Chichester, 2008; [Google Scholar]
- 1f. Seyferth D., Organometallics 2009, 28, 1598–1605. [Google Scholar]
- 2.
- 2a. Markies P. R., Schat G., Akkerman O. S., Bickelhaupt F., Smeets W. J. J., van der Sluis P., Spek A. L., J. Organomet. Chem. 1990, 393, 315–331; [Google Scholar]
- 2b. Markies P. R., Akkerman O. S., Bickelhaupt F., Smeets W. J. J., Spek A. L., Adv. Organomet. Chem. 1991, 32, 147–226; [Google Scholar]
- 2c. Holloway C. E., Melnik M., J. Organomet. Chem. 1994, 465, 1–63. [Google Scholar]
- 3.
- 3a. Bogdanović B., Maruthanuthu M., J. Organomet. Chem. 1984, 272, 115–122; [Google Scholar]
- 3b. Bogdanović B., Bons P., Konstantinović S., Schwickardi M., Westeppe U., Chem. Ber. 1993, 126, 1371–1383. [Google Scholar]
- 4.
- 4a. Lühder K., Nehls D., Madeja K., J. Prakt. Chem. 1983, 325, 1027–1029; [Google Scholar]
- 4b. [RMg(μ-R)2MgR], R: o,o′ -(C2H5)2C6H3: Wehmschulte R. J., Twamley B., Khan M. A., Inorg. Chem. 2001, 40, 6004–6008. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Schlenk W., W. Schlenk Jr. , Ber. Dtsch. Chem. Ges. 1929, 62, 920–924; [Google Scholar]
- 5b. W. Schlenk Jr. , Ber. Dtsch. Chem. Ges. 1931, 64, 734–736. [Google Scholar]
- 6.
- 6a.[{{o,o′-(iPr2C6H3)2C3H2N2}2MgCl(CH2SiMe3)}2(μ-dx)]: Danopoulos A. A., Braunstein P., Oil Gas Sci. Technol. 2016, 71, 24; [Google Scholar]
- 6b.[(Me3Si)2C{MgBr(η 1-dx)2}2]: Fischer R., Görls H., Langer J., Enke M., Westerhausen M., Organometallics 2016, 35, 587–594. See also: [Google Scholar]; Fischer R., Görls H., Krieck S., Westerhausen M., Z. Anorg. Allg. Chem. 2017, 643, 1276–1294. [Google Scholar]
- 7.R: Cy, Bz, Mes: Langer J., Krieck S., Fischer R., Görls H., Walther D., Westerhausen M., Organometallics 2009, 28, 5814–5820. [Google Scholar]
- 8.
- 8a.R: Et: Fischer R., Walther D., Gebhardt P., Görls H., Organometallics 2000, 19, 2532–2540; [Google Scholar]
- 8b.R: neo-Pent: Parvez M., Pajerski A. D., H. G. Richey, Jr. , Acta Crystallogr. Sect. C 1988, 44, 1212–1215; [Google Scholar]
- 8c.R: iPr: Blasberg F., Bolte M., Wagner M., Lerner H.-W., Organometallics 2012, 31, 1001–1005; [Google Scholar]
- 8d.R: Ph: Gärtner M., Fischer R., Langer J., Görls H., Walther D., Westerhausen M., Inorg. Chem. 2007, 46, 5118–5124. [DOI] [PubMed] [Google Scholar]
- 9. Clegg W., Brown D. A., Bryan S. J., Wade K., J. Organomet. Chem. 1987, 325, 39–46. [Google Scholar]
- 10. Jaenschke A., Olbrich F., Behrens U., Z. Anorg. Allg. Chem. 2009, 635, 2550–2557. [Google Scholar]
- 11. Jaenschke A., Paap J., Behrens U., Z. Anorg. Allg. Chem. 2008, 634, 461–469. [Google Scholar]
- 12. Gaderbauer W., Zirngast M., Baumgartner J., Marschner C., Tilley T. D., Organometallics 2006, 25, 2599–2606. [Google Scholar]
- 13. Baillie S. E., Clegg W., Garcia-Alvarez P., Hevia E., Kennedy A. R., Klett J., Russo L., Organometallics 2012, 31, 5131–5142. [Google Scholar]
- 14. Garcés A., Sanchez-Barba L. F., Fernandez-Baeza J., Otero A., Honrado M., Lara-Sanchez A., Rodrigez A. M., Inorg. Chem. 2013, 52, 12691–12701. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a.R=C{Si(CH3)3}3: Al-Juaid S. S., Eaborn C., Hitchcock P. B., Geary C. A. Mc, Smith J. D., J. Chem. Soc. Chem. Commun. 1989, 273–274; [Google Scholar]
- 15b.R=Mes*: Wehmschulte R. J., Power P. P., Organometallics 1995, 14, 3264–3267. [Google Scholar]
- 16. Stuhl C., Anwander R., Dalton Trans. 2018, 47, 12546–12552. [DOI] [PubMed] [Google Scholar]
- 17.Mg1⋅⋅⋅Mg1B in [R2Mg(μ-dx)]∞ with R=Et: 1173 pm, R=Ph: 1155.8 pm, R=Cy: 1240.4 pm, R=iPr: 1210.8 pm.
- 18.Mg1⋅⋅⋅Mg1A in [R2Mg(μ-dx)]∞ with R=Et: 655.1 pm, R=Ph: 656.49 pm, R=Cy: 690.4 pm, R=iPr: 686.6 pm.
- 19. Garcés A., Sachez-Barba L. F., Fernandez-Baeza J., Otero A., Lara-Sanchez A., Rodrigez A. M., Organometallics 2017, 36, 884–896. [Google Scholar]
- 20. Addison A. W., Rao T. N., Reedijk J., van Rijn J., Verschoor G. C., J. Chem. Soc. Dalton Trans. 1984, 1349–1356. [Google Scholar]
- 21. Waggoner K. M., Power P. P., Organometallics 1992, 11, 3209–3214. [Google Scholar]
- 22.[(Ph-C≡C)2Mg(thf)4]: Schwamm R. J., Coles M. P., Organometallics 2013, 32, 5277–5280. [Google Scholar]
- 23. Lichtenberg C., Spaniol T. P., Peckermann I., Hanusa T. P., Okuda J., J. Am. Chem. Soc. 2013, 135, 811–821. [DOI] [PubMed] [Google Scholar]
- 24. Sánchez-Barba L. F., Hughes D. L., Humphrey S. M., Bochmann M., Organometallics 2005, 24, 5329–5334. [Google Scholar]
- 25. Guino-o M. A., Baker E., Ruhlandt-Senge K., J. Coord. Chem. 2008, 61, 125–136. [Google Scholar]
- 26. Pérucaud M.-C., Le Bihan M.-T., Acta Crystallogr. Sect. B 1968, 24, 1502–1505. [Google Scholar]
- 27. Sarma R., Ramirez F., McKeever B., Chaw Y. F., Marecek J. F., Nierman D., McCaffrey T. M., J. Am. Chem. Soc. 1977, 99, 5289–5295. [Google Scholar]
- 28. Vestergren M., Eriksson J., Håkansson M., J. Organomet. Chem. 2003, 681, 215–224. [Google Scholar]
- 29. Metzler N., Nöth H., Schmidt M., Treitl A., Z. Naturforsch. Sect. B 1994, 49, 1448–1451. [Google Scholar]
- 30. Jäger E., Lehrwerk Chemie, Arbeitsbuch 5, Elektrolytgleichgewichte und Elektrochemie. VEB Deutscher Verlag für Grundstoffindustrie, Leipzig, 1977. [Google Scholar]
- 31. Fulmer G. R., Miller A. J. M., Sherden N. H., Gottlieb H. E., Nudelmann A., Stolz B. M., Bercaw J. E., Goldberg K. I., Organometallics 2010, 29, 2176–2179. [Google Scholar]
- 32.R. Hooft, COLLECT, Data Collection Software; B. V. Nonius, Netherlands, 1998.
- 33. Otwinowski Z., Minor W. in Macromolecular Crystallography, Part A, Methods in Enzymology, Vol. 276 (Eds.: C. W. Carter, R. M. Sweet), Academic Press, New York, 1997, pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 34.SADABS 2.10, Bruker-AXS Inc., 2002, Madison, WI, USA.
- 35. Sheldrick G. M., Acta Crystallogr. Sect. A 2008, 64, 112–122. [DOI] [PubMed] [Google Scholar]
- 36. Sheldrick G. M., Acta Crystallogr. Sect. C 2015, 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spek A. L., Acta Crystallogr. Sect. C 2015, 71, 9–18. [DOI] [PubMed] [Google Scholar]
- 38.XP, Siemens Analytical X-Ray Instruments Inc., Karlsruhe, Germany, 1990; Madison, WI, USA, 1994.
- 39.POV-Ray, Persistence of Vision Raytracer, Victoria, Australia, 2007.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary