

Abstract
Colorectal cancer is one of the so-called westernized diseases and the second leading cause of cancer death worldwide. On the basis of global epidemiological and scientific studies, evidence suggests that the risk of colorectal cancer is increased by processed and unprocessed meat consumption but suppressed by fibre, and that food composition affects colonic health and cancer risk via its effects on colonic microbial metabolism. The gut microbiota can ferment complex dietary residues that are resistant to digestion by enteric enzymes. This process provides energy for the microbiota but culminates in the release of short-chain fatty acids including butyrate, which are utilized for the metabolic needs of the colon and the body. Butyrate has a remarkable array of colonic health-promoting and antineoplastic properties: it is the preferred energy source for colonocytes, it maintains mucosal integrity and it suppresses inflammation and carcinogenesis through effects on immunity, gene expression and epigenetic modulation. Protein residues and fat-stimulated bile acids are also metabolized by the microbiota to inflammatory and/or carcinogenic metabolites, which increase the risk of neoplastic progression. This Review will discuss the mechanisms behind these microbial metabolite effects, which could be modified by diet to achieve the objective of preventing colorectal cancer in Western societies.
Colorectal cancer is the second most common cancer in women and the third in men worldwide, with slightly increased case numbers in men1. In 2012, 1,361,000 new cases were reported, which approximates to 10% of the total worldwide cancers — around half will die from the cancer1. The geographical variation in colorectal cancer incidence is remarkable, as illustrated by the latest WHO data1 (FIG. 1). Colorectal cancer is one of the so-called westernized diseases with the highest incidence rates in North America, Australia, New Zealand and Europe (all >40 cases per 100,000) and lowest in rural Africa (<5 cases per 100,000) and Asia (variable). However, at the extreme upper end of incidence rates are the Alaska Native people, with reported rates exceeding 100 cases per 100,000 (REF 2). This degree of variation, taken together with migration studies and experimental studies, which will be discussed, provide compelling evidence that environmental factors rather than genetic dysfunction are responsible for the development of colorectal cancer.
Figure 1 |. Geographical variation in colorectal cancer rates.
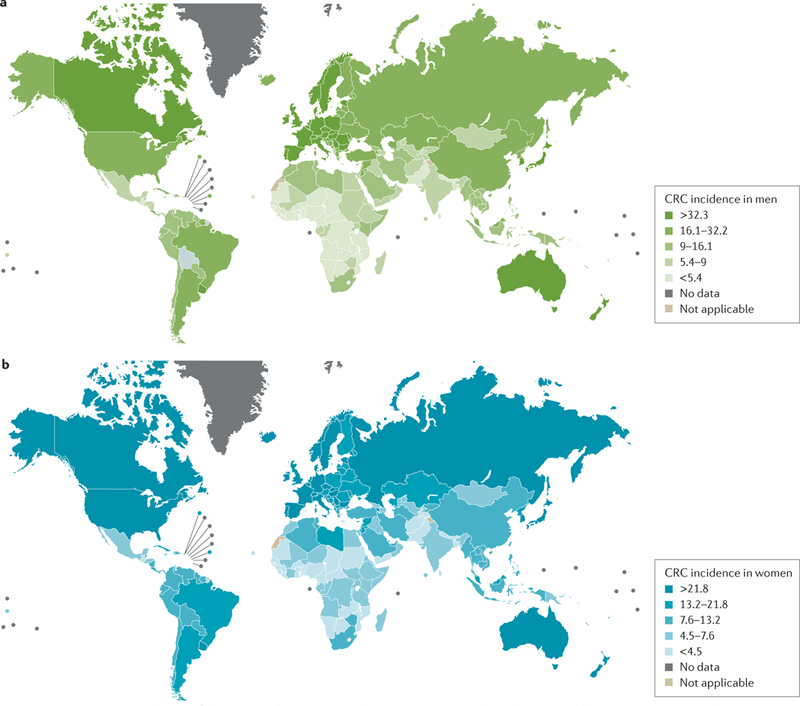
Data from the WHO1 demonstrating the high incidence of colorectal cancer (CRC) in westernized countries in a | men and b | women. Reprinted from GLOBOCAN 2012, v1.0, Ferlay, J. et al., Cancer incidence and mortality worldwide: International Agency for Research on Cancer CancerBase No. 11 http://globocan.iarc.fr/Pages/factsheetscancer.aspx?cancer=colorectal (2013).
Genome-wide association studies have identified 14 loci harbouring common variants that influence the risk of developing colorectal cancer3, but it took the inclusion of >9,000 cases and 9,000 controls before a statistically significant interaction between one of these loci and the intake of processed meat was found4. By contrast, significant associations between dietary factors and cancer risk have been demonstrated with much smaller numbers. The World Cancer Research Fund’s 2010 Continuous Update Report based on the systematic review and meta-analysis of 43 cohort or randomized controlled trials graded the evidence of linking dietary fibre with a decreased risk of colorectal cancer, and red and preserved meat with increased risk, as “convincing” — the strongest grade assigned5–7. Spurred by this analysis, the WHO issued a warning on the cancer risk of excessive consumption of meat and processed meat8. Other dietary factors, most notably fruit and vegetables, fish oils and calcium are associated with reduced risk6. Over three decades ago, Doll and Peto9 estimated from the epidemiological evidence of the time that >90% of gastrointestinal cancers were diet-driven. Although there is debate that this figure is too high for some gastrointestinal cancers — for example, in the stomach other environmental factors such as Helicobacter pylori have a role10 — experimental evidence will be provided to support the proposal that diet is the chief driver in the observed 20-fold geographical variation of sporadic colon cancer. The strength of environment is further illustrated by Le Marchand’s classic studies of Japanese immigrants to Hawaii who, within one generation, suffered a change in colon cancer incidence from the low rate of the Japanese native population to the high rate of Hawaiian natives11. The excitement of these analyses is that, unlike the cancer risk attributable to inherited genetic make-up, the risk due to these dietary factors is readily modifiable by the power of education and the modern media.
Mechanisms of colorectal cancer
As summarized by Nosho et al.12, colorectal carcinogenesis is a heterogeneous process with a differing set of somatic molecular alterations influenced by diet, environmental and microbial exposures and host immunity. However, not everybody who eats a high-meat, high-fat, low-fibre diet develops colorectal cancer, and not everybody who eats a balanced diet rich in fruits, vegetables and coarse grains is protected from the disease. The vast majority of colorectal cancers are not inherited but sporadic and what a person eats can have a profound effect on the initiation, promotion and progression of the neoplastic process. The rate of carcinogenesis is determined by the penetrance of the genetic defect and by the aggressiveness of the environmental insult. Evidence indicates that colorectal cancer arises from a stepwise disturbance of the composition of the gut microbiota, induced by food components or diet, plus genetic alterations in oncogenes and tumour-suppressor genes. For example, most colorectal cancers harbour mutations in the gene encoding APC, a tumour suppressor that regulates Wnt signalling by controlling the levels of β-catenin, which then regulates downstream inflammatory, cell cycle and proliferative pathways13. Mechanistic studies have shown that mutant Apc mice spontaneously develop adenomatous polyps14,15. However, experimental studies have shown that microbial fermentation products (such as butyrate) and microbial activated phytochemicals (such as polyphenols) have a wide range of antineoplastic effects that counteract tumorigenic signalling pathways and mount epigenetic mechanisms such as histone deacetylase inhibition that promote apoptosis, suppress proliferation and arrest neoplastic transformation16–22 (FIG. 2).
Figure 2 |. Importance of dietary residues and the colonic microbiota in determining colon cancer risk.
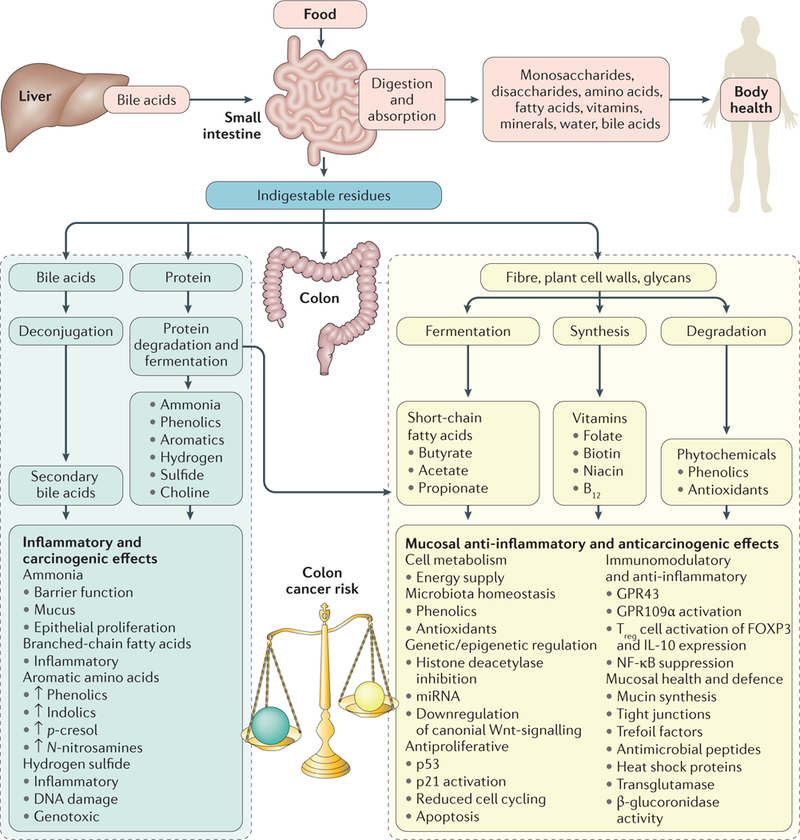
In health, >90% of a normal diet is absorbed in the small intestine and nutrients are distributed to maintain general body health. Residues entering the colon are chiefly complex carbohydrates (fibre), but also contain protein residues and primary bile acids secreted by the liver in response to fat ingestion. These residues have a critical role in the maintenance of colonic health as they determine the composition and metabolic activity of the colonic microbiota which, through fermentation, maintain mucosal and colonic health. With a balanced diet, saccharolytic fermentation of carbohydrates is dominant, producing short-chain fatty acids, particularly butyrate, which is the preferred energy source for colonocytes and has anti-inflammatory and antineoplastic properties through actions shown. With an imbalanced high-fat, high-meat, low-fibre diet, the proinflammatory and proneoplastic properties of protein fermentation and bile acid deconjugated residues predominate, leading to increased colon cancer risk.
Gut microbiota and colorectal cancer risk
Gut microbiota can influence the development of colorectal cancer in several ways. Inflammation is a necessary trigger but inflammation alone, or the presence of bacteria or bacterial metabolites alone, is not enough to promote tumorigenesis. Instead, complex inter-relationships with the gut microbiota, inflammation, host genetics and other environmental factors are needed for the progression of colorectal tumours23. Microorganisms can induce chronic inflammation in a number of ways: adherence to the epithelium; activation of an immune response through binding to Toll-like receptors and/or activating regulatory T (Treg)cells; synthesis and secretion of cytotoxic biomolecules or metabolites; or by translocation into the body24,25. The critical role of the gut microbiota in colorectal cancer is shown by the association between microbial abundance and cancer incidence. For example, neoplastic transformation is most common in the distal gut where the microbiota are most dense, and carcinogenesis in experimental animals is linked to the presence or absence of microorganisms26,27. Although great effort has been expended searching for a single causative organism — as in H. pylori-associated gastric ulceration and cancer — the evidence to date points to a shift in composition and activity of the gut microbiome, creating a microclimate that promotes inflammation, proliferation and neoplastic progression. For example, a review of 31 original articles on the role of colon microbiota in colorectal cancer performed in humans and animals observed that, despite differences in methodology, some bacteria were consistently augmented (including Fusobacteria, Alistipes, Porphyromonadaceae, Coriobacteridae, Staphylococcaceae, Akkermansia spp. and Methanobacteriales), whereas others were consistently diminished in colorectal cancer (Bifidobacterium, Lactobacillus, Ruminococcus, Faecalibacterium spp., Roseburia and Treponema)28. In addition, levels of some microbial metabolites (such as nitrogenous compounds) were consistently elevated, whereas others (such as butyrate) were decreased throughout colonic carcinogenesis28.
In another study, faecal samples from three patients with colorectal cancer and three healthy individuals as controls were transplanted into germ-free mice in which cancer was chemically promoted29. The rate of tumour generation was associated with microbial composition. The taxa most positively associated with tumour burden were members of Gram-negative Bacteroides, Parabacteroides, Alistipes and Akkermansia, whereas Gram-positive Clostridiales, including multiple members of Clostridium group XlVa, were negatively correlated with tumours29. Furthermore, metagenomic analysis inferred a negative correlation between tumour count and the potential for butyrate production, and a positive correlation between tumour count and the capacity for host glycan degradation, suggesting a role for mucus degraders in tumorigenesis29.
The application of the compositional pattern of the faecal microbiota in the prediction of adenoma and carcinoma has also been examined in 60 patients and 30 healthy controls30. This analysis revealed both an enrichment and depletion of several bacterial populations associated with adenomas and carcinomas. Combined with known clinical risk factors of colorectal cancer (such as BMI, age and race), data from the gut microbiome improved the ability to differentiate between healthy, adenoma and carcinoma clinical groups relative to risk factors by 50-fold.
Similar conclusions were reported by a European study based on faecal samples from a mixed sample of healthy individuals (colonoscopy negative), patients with adenoma, and patients with confirmed colorectal cancer from France, Germany, Denmark and Spain31. The Gram-negative phyla of Fusobacteria and, to a lesser extent, Proteobacteria were substantially increased in patients with cancer, whereas Gram-positive Actino bacteria were decreased31. Bacteroidetes and Firmicutes were enriched and depleted, respectively, in patients with colorectal cancer. In addition, meta-genomic sequencing of faecal samples was used to identify taxonomic markers that distinguished patients with colorectal cancer from tumour-free controls and found that pattern differences were able to predict colorectal cancer at a sensitivity similar to that of the usually used faecal occult blood test31. When the two tests were combined, detection sensitivity increased by 45%. On the basis of these findings, carcinogenesis was proposed to result from a metabolic shift of the gut microbiota from the normal fibre degradation to utilization of host carbohydrates and amino acids, accompanied by an increase in lipopolysaccharide metabolism.
Despite the substantial support for carcinogenesis being linked to an overall disturbance in microbial balance and integration, accumulating evidence suggests a predominant role of Fusobacterium spp. in the neoplastic process; biopsy material from adenomas and adjacent normal mucosa in 19 patients showed 48% positivity for Fusobacterium in tumour tissue, which was higher than in the normal tissue32. Using the ApcMin/+ mouse model of intestinal tumorigenesis, Fusobacterium nucleatum was shown to increase tumour multiplicity and recruit tumour-infiltrating myeloid cells that can promote tumour progression32. Other studies have shown that F. nucleatum-high colorectal cancer tissues were inversely associated with the density of CD3+ T cells33 and had a strong association with microsatellite instability and CpG island methylator phenotype-linked cancers34. These examples demonstrate the complex interactions between microorganisms, immunity, genetic predisposition and colorectal cancer.
Also noteworthy is the evidence for the potential of individual microorganisms to induce colitis and colorectal cancer in experimental animals35. The microorganismstudied was the human commensal enterotoxigenic Bacteroides fragilis (ETBF), a subset of Bacteroides fragilis distinguished by the secretion of a specific enterotoxin, which has been associated with ulcerative colitis and colorectal cancer in humans36. First, ETBF infection in mice was shown to cause a brief, acute colitis characterized by rapid and robust colonic activation of STAT3, which induced a type 17 T helper cell immune response and evolved into chronic colitis35. Second, ETBF colonization of multiple intestinal neoplasia in mice induced numerous IL-17A-dependent distal colon adenomas37. Third, the persistence of the ETBF infection has been shown to be essential for tumour formation, as antibiotic therapy prevented tumorigenesis37. Finally, fascinating evidence is emerging on the ability of gut microorganisms to cross the mucosal or blood barrier and establish unique micro-biomes within solid body organs, such as breast tissue38, seminal vesicles39 and placenta40. These findings might explain the association between dysbiosis and the use of antibiotics and the increased risk of cancers of the colon, gastric, oesophageal, pancreatic, laryngeal, breast and gallbladder41. In a qualitative survey of the breast microbiota DNA, the bacterium Methylobacterium radiotolerans was relatively enriched in tumour tissue, whereas the bacterium Sphingomonas yanoikuyae was enriched in paired normal tissue42. Further research is needed to ascertain whether these microorganisms can influence carcinogenesis by direct attachment to organ cells or whether it is through the production of toxic metabolites.
The burning question remains whether the discussed microbial associations with cancer are cause or effect. For example, tumorigenesis might produce inflammation, ulceration and necrosis, changing the microenvironment and the growth conditions for different microorganisms. For the remainder of this Review, the experimental evidence supporting a major role of microbial metabolites in initiating, promoting and advancing colorectal cancer will be examined.
The role of diet
Food is a complex mixture of thousands of bioactive molecules, many of which are modified by preservation, cooking methods, digestion, metabolism by the host and the luminal gut microbiota. Although evidence suggests that an excess of nutrients such as protein and fat might be inflammatory and therefore might potentiate neoplastic change43, the evolutionary interaction between food and genes is in the preservation of mucosal health — from what we know of the process of evolution, the dietary needs of every organism are genetically determined. Some have argued that our health will be better served by the diet that first established Homo sapiens in the Palaeolithic era in Africa, a period that lasted from ~2.5 million to 11,000 years ago44,45. With regard to the colon, an ideal microbiota probably evolved in tandem with the development of our colon. Although the changes in diet following the Industrial Revolution provided more food for growth, the diet became progressively depleted in colonic food, namely fibre, leading to a shift in gut microbiota metabolism that does not provide for colonic mucosal health.
Advances in dental microwear and stable isotope technology have shed light on the composition of foods consumed by early hominins (Australopithecus) and our Homo ancestors46, providing evidence that dietary consumption was diverse, from Paranthropus robustus consuming fleshy fruits and soft young leaves (browsers), to P. boisei living on a diet rich in tropical grasses and sedges (grazers). Coprolite analyses from prehistoric deposits in caves in Southwest Arizona dating back 14,000 years47 have indicated that the diet of early Homo sapiens was predominantly high in complex carbohydrate and fibre. Furthermore, dental microwear and stable isotope analyses have hinted at unexpected diversity and complexity in early hominin diets, but have concluded that their diets were most likely dominated by plant foods48. Although these analyses provide no direct information on the quantity of meat and fat consumed, they do suggest that the diet throughout our evolution has been consistently rich in high-fibre foods. Thus, it is reasonable to propose that the appearance of colorectal cancer as one ofthe so-called westernized diseases is a consequence of the progressive diminution in the consumption of fibre-rich foods and the subsequent dysbiosis of our gut microbiota. Studies49–51 have shown that colorectal cancer remains rare in semi-urbanized agricultural-commercial communities in Africa, who continue to consume >50 g of fibre per day, indicating that we do not have to return to hunter-gatherer status to avoid colorectal cancer, we just need to pay more attention to the nutritional needs of the colon. The concern is that our extraordinary ability to adapt to the environment and increase our lifespan has not enabled us to identify the phenotypic identity of colorectal cancer until it is too late.
Colonic nutrition
As the undigested dietary residues that enter the colon and provide an energy source for the microbiota are chiefly complex carbohydrate fibres, mutualism will be best served by a diet containing sufficient fibre. The fermentation of fibre releases short-chain fatty acids (SCFAs), principally butyrate, propionate and acetate, which are essential for colonic mucosal health and otherwise unavailable from the diet (small quantities might be ingested but are absorbed by the small intestine). Much of the original work on the ability of the colonic microbiota to ferment fibre for their own metabolic needs, with the subsequent production of luminal SCFAs, was performed by the laboratories of Macfarlane52,53, Cummings54 and Englyst54. Roediger was the first to demonstrate that butyrate and not glucose was the preferred energy source of colonocytes, thus establishing the foundation for the now well-recognized symbiotic relationship between colonic microorganisms and colonic mucosal health55.
Saccharolytic fermentation
Fermentation of complex carbohydrate or starch involves numerous intermediate steps and multiple enzyme pathways contributed by different microorganisms45,46 (FIG. 3). The initial fermentation of carbohydratethat escaped digestion in the small intestine is followed by the utilization and cross-sharing of metabolites by different members of the microbiota, then finally the synthesis of SCFAs56. The most abundant SCFAs are butyrate, propionate and acetate. Although the molar ratios of these SCFAs vary with the composition of the microbial fermenters and the fibre source, the relative magnitude of the quantities are acetate > propionate > butyrate, which is important in considering the role each of the SCFAs has on mucosal and body homeostasis57,58. Reducing the carbohydrate content of the diet from the usual 52% to 4%, and increasing the protein content from 13% to 30%, caused a reduction in the proportion of butyrate measured in faecal samples from 16% to 8% in patients with obesity59. This finding was associated with a change in microbiota composition, with a reduction in the abundance of butyrate-producing bacteria related to Roseburia spp. and Eubacterium rectale.
Figure 3 |. Key metabolic pathways of the colonic microbiota.
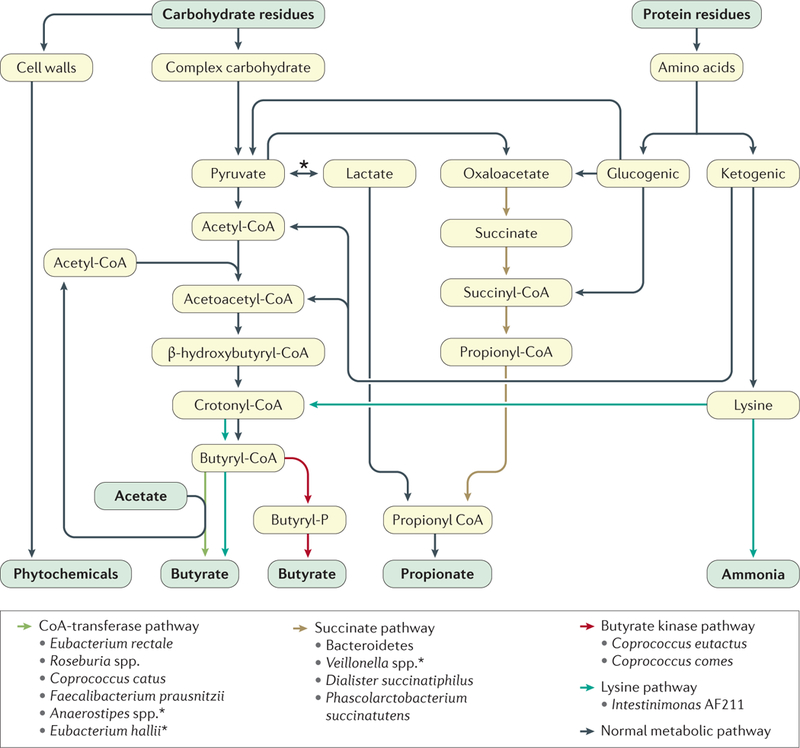
Through the process of fermentation, these metabolic pathways culminate with the production and release of the major short-chain fatty acids (acetate, propionate and butyrate) and phytochemicals from plant cell walls56. Note that there are two microbial enzymes that are responsible for the final synthesis of butyrate, namely butyryl-CoA transferase (dominant, formed by a variety of genera and species) and butyrate kinase (favoured in proteolytic fermentation). Also shown is the pathway for converting the amino acid lysine into butyrate, which also generates ammonia69. The main propionate production pathway is the succinate pathway. Bacterial species found to use certain pathways are shown but are not exhaustive. The asterix indicates species shown to use lactate to form butyrate56.
Although butyrate is unique in its role as the chief energy source for the colonic mucosa, acetate and propionate share many of the same anti-inflammatory and antineoplastic properties of butyrate60. Furthermore, because acetate and propionate are less metabolized by the mucosa, more is absorbed to have systemic effects that influence weight gain, obesity and the metabolic syndrome61. Importantly, acetate suppresses appetite and enhances lipogenesis62, and propionate suppresses cholesterol synthesis63–65. Quantitatively, saccharolytic fermentation usually salvages ~150–250 kcal per day, but in malabsorptive conditions such as the short bowel syndrome, it can salvage up to ~1,000 kcal per day66.
Proteolytic fermentation
Quantitatively, proteolytic fermentation is smaller than saccharolytic fermentation, as protein digestion and absorption by the small intestine is more efficient (>95%67) and the quantities of protein consumed in the diet are generally lower than those for carbohydrate, unless diets such as the Atkins or Banting diet are being followed. However, there is a substantial endogenous supply of protein derived from epithelial desquamation, secretions and mucus, and microorganisms have an important role in recycling body nitrogen and amino acids68. Protein fermentation differs from saccharolytic fermentation in that, in addition to producing many of the same SCFAs from the carbon skeleton, it also releases potentially toxic nitrogenous and sulfur metabolites, such as ammonia, amines, nitrates, nitrites and hydrogen sulfide68. One study suggested that protein fermentation might have health benefits, demonstrated by the presence of a unique bacterium, Intestinimonas strain AF211, belonging to Clostridium cluster IV (Lachnospiraceae) of the Firmicutes phylum in 50% of healthy volunteers69. Culture, isotope labelling and metagenomic analysis indicated that this microorganism contained all the enzymatic pathways for converting lysine into butyrate. However, this catabolic pathway also generated ammonia.
Ammonia can be absorbed and recycled in transamination pathways in the liver and can be taken up and detoxified by probiotic Lactobacilli; however, in high concentrations, ammonia interferes with cell metabolism, is cytotoxic and induces inflammation and epithelial proliferation70. In rats, it was shown that ammonia interacted with N-nitroso compounds to induce colorectal cancer71.
Many of the protein fermentation metabolites can be taken up by other microorganisms and synthesized into activated carcinogens. For example, amines and nitrates can be used by facultative and anaerobic colonic bacteria to catalyse the formation of N-nitrosamines, which are among the most potent experimental procarcinogens72,73. The clinical relevance of this finding was shown in a study in which eight healthy volunteers were assigned a diet containing 60 g or 600 g meat in a randomized crossover study. Those on the high-meat diet had threefold higher faecal concentrations of nitrosamines than those who consumed a low-meat diet74.
Hydrogen sulfide is produced by sulfate-reducing bacteria in response to sulfur compounds derived from the diet, intestinal secretions and cell desquamation, or sulfur-rich bile acids such as taurine. Experimental studies have concluded that hydrogen sulfide generated by these microorganisms is pro-inflammatory75 and genotoxic at physiological concentrations76. However, exciting new studies in rats have shown that endogenous mucosal hydrogen sulfide increases ulcer healing and is anti-inflammatory77,78. Furthermore, the same investigative team demonstrated that a hydrogen-sulfide-releasing form of the NSAID naproxen was more effective than naproxen alone in suppressing tumour formation in the azoxymethane (AOM)-induced mouse intestinal adenoma model79. This finding opens up a new, potentially safer approach to colon cancer chemoprevention than aspirin, without the added risks of gastrointestinal bleeding. These divergent findings might be explained by confusing association with cause. For example, is the finding of increased colonic hydrogen sulfide production or defective detoxification of hydrogen sulfide in ulcerative colitis80 an appropriate finding after all, possibly representing an enhanced drive towards ulcer healing rather than the cause of inflammation? Clearly, more work is needed in this area.
Protein fermentation also results in the release of aromatic amino acids, which in turn are metabolized to phenols and p-cresol before being excreted in the urine as potential biomarkers of protein consumption68. Although the evidence for their carcinogenic potential in humans is limited, experimental studies have shown an ability to damage cellular structure and increase permeability81. Theoretically, these changes could increase the susceptibility of the colonic epithelium to luminal carcinogens. Despite these experimental findings, there is no evidence that high-protein diets increase the risk of colon cancer in humans. This finding is probably explained by the fact that protein is never eaten on its own and that the other components of the diet outweigh any deleterious effects protein metabolites might have on mucosal health68. For example, studies in humans have shown that fibre supplementation reduces feacal ammonia and p-cresol82, and increases the colonic microbial uptake of nitrogenous metabolites83. Additionally, the administration of synbiotics in a variety of human studies consistently reduced protein fermentation products and the genotoxicity of faecal water68. Furthermore, symbiotic supplementation (Lactobacillus rhamnosus GG, Bifidobacterium lactis BB12 and inulin) was associated with less DNA damage and reduced mucosal proliferation rates in patients who had undergone colonic polypectomy84, supporting the view that cancer risk is determined by the balance between mucosa health-promoting and inflammatory or carcinogenic precursors. Strong evidence (further discussed in a following section) also suggests that the metabolic and carcinogenic effects of high-meat diets can be ameliorated by the simultaneous consumption of fibre in the form of resistant starch85.
Butyrate
Diminished faecal butyrate levels might not only be a biomarker of cancer risk, but also cancer progression and severity22. Clinical studies have revealed that, compared with healthy controls, individuals with advanced colorectal cancer have diminished butyrate-producing bacteria and lower levels of SCFAs86–88.
A wealth of experimental evidence (reviewed else-where24,25,89) has demonstrated the inhibitory effect of butyrate on tumorigenesis through multiple mechanisms (FIG. 2). Critical actions include its anti-inflammatory and immunomodulatory effects90,91, downregulation of the key canonical Wnt signalling pathway linked to colonic carcinogenesis15, inhibition of proliferation and migration of neoplastic cells, restriction of tumour angiogenesis, induction of apoptosis and the promoted differentiation of neoplastic colonocytes92–98. Butyrate has an important role in reinforcing the mucosal defence barrier by enhancing the expression of mucin-encoding genes and the induction of trefoil factors, heat shock proteins, antimicrobial peptides and transglutaminase activity99–102. Finally, evidence also exists that the powerful actions of butyrate on the maintenance of colonic mucosal health are at least partly mediated via the colonic Treg-cell activation of FOXP3 and IL-10 expression103. In an elegant series of studies based on specific pathogen-free mice, gnotobiotic altered Schaedler-floracolonized mice and germ-free mice, SCFA was shown to regulate the size and function of the colonic Treg pool103. This process protected against colitis, possibly through interactions between free fatty acid receptor 2 (also known as G-protein coupled receptor 43 or GPR43) induction, the direct activation of histone deacetylase inhibition, and upregulation of IL-10 gene expression.
In one clinical study, the 5-year history of fibre consumption and faecal SCFA concentrations in 344 patients with advanced adenomas was compared to 344 healthy, aged-matched individuals as controls87. Fibre intake was significantly (P <0.05) reduced in patients with colorectal adenoma, as were levels of faecal SCFAs. Microbiota composition and quantities were measured by pyro-sequencing and PCR in 47 volunteers from each group, and principal component analysis showed distinct differences in the faecal microbiota communities of the two groups. Clostridium, Roseburia and Eubacterium spp. were significantly (P <0.001) less prevalent in the advanced adenoma group than in healthy individuals, whereas Enterococcus and Streptococcus spp. were more prevalent in the adenoma group. Interestingly, butyrate and recognized high-butyrate-producing bacteria were more prevalent in healthy controls consuming a high-fibre diet than in patients with colorectal adenoma consuming a high-fibre diet, suggesting colonic butyrate deficiency due to low-fibre intake or diminished butyrate-producing bacteria promotes neoplasia.
Investigating mechanisms linking fibre intake to neoplasia, a pathway involving oncogenic microRNA (miRNA; oncogenic miR-17–92a cluster) biogenesis was unravelled, through which butyrate might suppress the proliferation of human cancer cell lines86,104. First, miR-92a levels were established as sevenfold higher in sporadic human colorectal cancer tissue than in adjacent normal tissue. Second, the addition of butyrate to a human cancer cell line was found to reduce the levels of primary miR-17–92a, precursor and mature miR-92a. This finding was associated with inhibition of the key MYC oncogene and enhanced CDKN1C (also known as p57) expression, causing suppression of proliferation and an increase in apoptosis of cancer cells, respectively. The same effect was produced by the addition of other histone deacetylase inhibitors, namely suberoylanilide hydroxamic acid and valproic acid, indicating that the mechanism was principally epigenetic. An increasing number of experimental and human studies have supported the suggestion that the chief mechanism behind the ability of butyrate to suppress proliferation and cancer risk is its capacity to normalize the function of oncologic miRNA clusters (oncomirs)85,105–107. For example, in one randomized controlled crossover study, healthy volunteers were given either a diet high in red meat (300 g per day) or a diet high in red meat plus a resistant starch (fibre) supplement for 4 weeks85. The diet high in red meat increased expression of oncogenic miRNA (miR-17–92 cluster) in rectal mucosa and cell proliferation, in association with a decrease in miR-17–92 target gene transcript levels85. Remarkably, all these effects were negated by increasing butyrogenesis through fibre supplementation (40 g of butyrylated high-amylose-maize starch per day, of which 60% is resistant).
Experimental evidence for an alternative carcinogenesis mechanism has been provided, based on the ability of SCFAs produced from fibre fermentation to bind to the SCFA receptor GPR43 in the colon22. In the AOM-dextran sodium sulfate (DSS) colitis-associated colon cancer rat model, a supplementation of the diet with resistant starch decreased tumour multiplicity and adenocarcinoma formation22. Evidence that the mechanism involved increased butyrogenesis was provided by the associated increases in microbial butyrate-producers (Ruminococcus and Bifidobacterium) and increased SCFA production. Furthermore, the increased butyro-genesis was associated with increased expression of the SCFA receptor GPR43 mRNA, which triggered reduced inflammation via inhibition of cyclooxygenase (COX)-2, nuclear factor-κB (NF-κB), TNF and IL-1β expression, as well as cell proliferation22.
A threshold effect of butyrate.
That butyrate can paradoxically stimulate mucosal proliferation under certain conditions has long been recognized. This effect can be explained by the mixed action of butyrate on cell growth: as the chief energy supply it will stimulate growth, as an antineoplastic agent it will suppress growth in a phenomenon termed the ‘butyrate paradox’. Evidence for the paradox includes the observation that increased butyrogenesis stimulates epithelial growth through its energy provision under starved or atrophic conditions, whereas enhanced butryogenesis under conditions of excess growth suppresses proliferation and cancer risk108. Experimental studies have shed light on the mechanisms behind the paradox, reporting that the quantity of butyrate produced might be critical. Low colonic crypt concentrations of butyrate (0.5 mM) had no histone deacetylase inhibitory effect because all the butyrate was used for cellular energetics, whereas the excess from higher quantities (5 mM) was able to spill into the nucleus and act as a histone deacetylase inhibitor, increasing differentiation and apoptosis, and suppressing proliferation of cancerous colonocytes109. Furthermore, in gnotobiotic mouse models colonized with wild-type or mutant strains of a butyrate-producing bacterium, fibre was demonstrated to have a potent tumour-suppressive effect but in a microbiota and butyrate-dependent manner110. However, it is not surprising to find studies that do not fit well with the idea that butyro-genesis suppresses carcinogenesis. For example, one study showed that a low-carbohydrate diet reduced key luminal butyrate-producing microorganisms (such as Clostridia cluster XIVa) and butyrate production in a colon cancer mouse model111. Surprisingly, this finding was associated with a reduction of colon cancer development. Backed up by cell culture studies, the authors proposed that the gut microbiota stimulate colorectal cancer by providing butyrate that drives aberrant proliferation or β-catenin activity, increasing the risk of neoplastic transformation. However, what the metabolic requirements of the colonic epithelium are in this nonphysio-logical model, and whether they were met or exceeded, is unknown.
Good evidence that excessive fermentation rates are as harmful as low rates also exists. In goats, a highgrain (65%) diet has been shown to result in sloughing of the surface layer epithelium, intercellular tight junction erosion, cell mitochondrial damage and upregulation of IL-2 and IFNγ mRNA expression in the colonic mucosa. These effects were in association with increased microbial fermenters, increased acidity and increased SCFA production112. The condition is also well recognized in cattle when dietary, animal or environmental factors contribute to abnormal, excessive flow of fermentable carbohydrates from the small intestine into the colon, resulting in distention, diarrhoea and severe mucosal injury113. Furthermore, it is important to note that humans also can have a similar condition. Massive loss of the small intestine, a condition termed short bowel syndrome, results in excessive carbohydrate delivery to the colon. In this condition, the colonic microbiota perform an essential function of increasing fermentation with salvage of up to ~1,000 carbohydrate calories per day66. However, disturbance of the microbiota composition can lead to an overgrowth of D-lactate-producing organisms and potentially lethal D-lactic acidosis, as this form of lactate cannot be metabolized by humans114.
The importance of a balanced diet
From the evidence discussed, and the mechanisms summarized in FIG. 2, the emerging picture is one in which the gut microbiota behave as a community wherein intermicrobial interactions strive to produce a metabolic phenotype that supports colonic health and function. From evolutionary considerations, the gut microbiota have a genetically determined need for food residues derived from a healthy balanced diet.
Provision of an imbalanced diet leads to disturbance in structure and function of the gut microbiota, with unopposed production of metabolites that can induce inflammation and proliferation that increase the risk of neoplasia. Critically, the inflammatory and proliferative effects induced by meat and bile acids, as well as the associated DNA damage and mutations, can be prevented by the simultaneous consumption of resistant starch85,107,115–119. This finding makes teleological sense as we are all omnivores.
Phytochemicals
Phytochemicals are a diverse group of micronutrients that are synthesized by plants and have remarkable antioxidant and antineoplastic effects in experimental models. Together with fibre, they account for the anticarcinogenic properties of fruits, vegetables and grains, or high-fibre foods. Studies on fermentation products have shown that their antineoplastic effects are additive to those of SCFAs16,120. As they are complex glycosylated molecules, considered xenobiotics and mostly stored within plant cells, phytochemicals have little influence on the upper gastrointestinal tract with 95% unabsorbed by the small bowel and entering the colon121. Within the colon, microbial fermentation releases these compounds and specific bacteria metabolize many of them into more bioactive metabolites. This process results in the accumulation of metabolites in the millimolar range at the surface of the mucosa, amplifying biological potency121. A good example of the relationship between plant-cell breakdown, fibre metabolism and phytochemical release is given by a study that reported that the feeding of a high-protein, low-carbohydrate and low-fibre diet to human volunteers resulted in reduced levels of key butyrate-producers (Roseburia and Eubacterium rectale), a decrease in the proportion of butyrate in faecal SCFA concentrations, and greatly reduced concentrations of free phenolic acids in the gut122. A forward and backward relationship exists between the metabolites and the gut microbiota, such that the specific microorganisms that metabolize phytochemicals, such as Escherichia coli, Bifidobacterium sp., Lactobacillus sp., Bacteroides sp. and Eubacterium sp. are stimulated by the increased concentrations of phytochemical metabolites121,123. As different polyphenols affect different bacteria, their effect on the gut microbiota composition is variable. The ability of phytochemicals to suppress Gram-positive bacteria could be due to the ability of the metabolites to bind to membranes of specific bacteria and trigger the release of hydrogen peroxide, which induces cell-wall breakdown and lysis124,125.
A wealth of experimental studies (reviewed elsewhere120,121,126) have shown that polyphenols, particularly those derived from grapes, tea, coffee and cocoa or chocolate, have powerful anti-inflammatory, antiproliferative and tumour-inhibiting properties in the colon. Mechanistic studies have suggested that these actions are mediated by modification of eicosanoid synthesis, downregulation of the inflammatory cascade (COX-2, NF-κB, AP-1, TNF, IL-6 and VEGF), regulation of DNA synthesis, and induction of luminal detoxifying enzymes (β-glucuronidase, β-glucosidase, β-galactosidase, mucinase and nitroreductase). In this way, phytochemicals complement many of the actions of SCFAs illustrated in FIG. 3, strengthening support for a balanced diet containing high-fibre fruits and vegetables.
This synergism explains why the effects of total saccharolytic fermentation products on cell proliferation were greater than that due to SCFAs alone16,120. Unfortunately, because of practical difficulties in measuring mucosal levels of phytonutrients in humans, there are few physiological studies on the relative effects of phytonutrients on mucosal metabolism and cancer risk. Estimates that have been obtained are generally based on metabolites in urine124. For example, a randomized controlled trial of dates (seven per day for 3 weeks), a rich source of fibre and polyphenols, showed a reduction in the genotoxicity of faecal water in normal healthy individuals in the absence of changes in specific faecal microbiota (including bifidobacteria and high butyrate-producers) and in SCFAs127. This finding suggests that either the observed increase in gut transit masked SCFA changes, or that polyphenols were more responsible for the antineoplastic changes. Unfortunately, no biological measurements of polyphenol metabolism were made and so the mechanism remains unexplained.
In the AOM-DSS rat model of colitis-associated colorectal cancer, the protective effects of resistant starch on adenoma and carcinoma formation were not seen when a high-polyphenol-containing green tea supplement (main component epigallocatechin-3-gallate) was added to the diet instead of resistant starch22. The dose of green tea given was estimated to be equivalent to 3–4 cups per day but, again, no biological measurements were made. In cell cultures, the inhibitory effects of polyphenols have been well demonstrated. For example, the ability of polyphenols and their metabolites, namely ellagitannin metabolites, at concentrations found in the colon following the consumption of pomegranates and walnuts, were shown to have strong antiproliferative effects in cancer cell lines. Using Caco-2 cells and primary tumour cells from a patient with colon cancer, mixtures of ellagitannin metabolites, ellagic acid and gut-microbiota-derived urolithins reduced the number and size of colonospheres and aldehyde dehydrogenase activity, indicating the potential for phytochemicals to control colon cancer chemoresistance and relapse128. This finding followed earlier studies reporting that urolithin metabolites were anti-inflammatory in human colonic fibroblasts129 and strongly antiproliferative in human colorectal cancer (Caco-2) and normal (CCD18-Co) cell lines130.
The antineoplastic potential of black raspberries has been studied for many years131,132, identifying anthocyanins (cyanidin-3-O-glucoside, cyanidin-3-O-rutinoside and cyanidin 3-O-2G-xylosylrutinoside) as the most potent anticarcinogenic constituents, whereas ellagitannins have been identified as the key components of tumour suppression in red raspberries. These studies have shown that extracts suppressed colon tumour formation by up to 70% in the AOM rat cancer model. Studies in tumorigenic cell lines suggest that the mechanism involves the inhibition of proliferation, induction of apoptosis and the modulation of the expression of COX-2 and inducible nitric oxide synthase132. Randomized controlled trials of black raspberries are now being performed in patients with a high risk of colon and oesophageal cancer. Preliminary findings of a phase 1b controlled clinical trial in 14 patients with familial adenomatous polyposis have been published, showing that supplementation of the diet for 9 months with 100 g black raspberry powder significantly (P <0.05) reduced the polyp burden and decreased staining of the mucosal proliferation biomarker Ki-67, compared with seven patients given placebo131.
Often the results of food studies on health produce disappointing results that cause confusion and dismay among the public, such as the effects of saturated fat and red meat on colorectal cancer and heart disease. Thus, it comes as a pleasant surprise to hear of the potential benefits of coffee and red wine on cancer and health. In a randomized crossover study on the effects of red wine (272 ml per day), nonalcoholic red wine (273 ml per day), or gin (100 ml per day) performed for 4 weeks, the microbiota, polyphenol and blood lipid responses in healthy volunteers were examined. Both red wine and nonalcoholic red wine significantly (P <0.05) increased the number of Enterococcus, Prevotella, Bacteroides, Bifidobacterium, Bacteroides uniformis, Eggerthella lenta, and Blautia coccoides-Eubacterium rectale groups, whereas gin had no effect133. Evidence that the changes were a consequence of increased polyphenol metabolism was provided by the observation that resveratrol metabolites derived from phase II metabolism were significantly (P <0.05) increased in 24 h urine collections after both red wines, compared with baseline and after gin intake. Furthermore, the concentration of dihydroresveratrol produced by the intestinal microbiota was also significantly (P <0.05) increased in the urine. Unfortunately, colonic saccharolytic fermentation products were not measured, nor were mucosal biomarkers of cancer risk. However, potential health benefits were shown as the microbiota changes were accompanied by reductions in levels of systolic and diastolic blood pressures, plasma triglyceride, total cholesterol, high-density lipoprotein cholesterol and C-reactive protein.
Although conflicting reports exist134,135, studies report an inverse association between coffee consumption and colorectal cancer, with high coffee consumers, in particular, showing reduced risk. For example, in a study from Japan with 463 cases of colon cancer and 340 cases of rectal cancer, the multivariate-adjusted odds ratio of colon cancer was found to significantly (P trend=0.02) decrease with increasing intakes of coffee polyphenols136. The association with distal colon cancer was strongest, and no statistically significant association with tea consumption was seen. A further study from the Middle East in 5,145 colorectal cancer cases and 4,097 healthy controls also showed an inversely associated risk of colorectal cancer with coffee consumption in a dose-dependent manner137. Compared with <1 serving of coffee per day, an intake of 1 to >2.5 servings per day was associated with significantly (P trend <0.001) lower odds of colorectal cancer. The dose-response trend was significant for both colon and rectal cancers. In a case-controlled study of 251 colorectal cancer cases and 247 cancer-free matched individuals as controls from the Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial, an untargeted metabolomic analysis of serum was performed and integrated with dietary analysis and colorectal cancer development138. The researchers identified three caffeine-related metabolites — theophylline, caffeine and paraxanthine — that were significantly associated with the absence of colorectal cancer development. Evidence also suggests that colon cancer progression in humans can be retarded by high coffee consumption135.
In summary, strong supportive evidence suggests that colorectal cancer suppression induced by a diet high in fruits, vegetables and grains is a consequence of, not only the high fibre content, but also the polyphenol content. This finding argues for the use of whole-foods rather than fibre supplements in the campaign to reduce the excessive rates of colorectal cancer in westernized societies.
Fat-stimulated metabolites
Epidemiological studies have shown that the relationship between dietary fat intake and colorectal cancer is not as strong as it is for meat and fibre. This finding might be explained by the weakness of questionnaires being able to segregate the effects of meat and energy from fat, as a high-meat diet is usually accompanied by a high intake of saturated fat. For example, in the Nurses’ Health Study, animal fat intake was positively associated with colon cancer incidence in women, although fat intake was not independent of total energy intake in the study139. A further explanation might involve the modulating effects of luminal metabolites derived from calcium and fibre140. Further confusion has arisen from experimental findings showing that saturated fat protected against colon cancer development in the AOM mouse model, which was attributed to the ability of saturated fat to stimulate mucin synthesis, thereby increasing intestinal barrier integrity141. In an attempt to overcome the weaknesses of dietary recall, the relationship between colorectal cancer occurrence and blood biomarkers of fat intake, namely plasma phospholipid fatty acids, was examined in 4,205 high-risk Australian participants, which included 395 colorectal cancer cases142. The results showed no association between dietary fat intake by questionnaire and colorectal cancer, but there was a significant (P <0.005) association between saturated plasma phospholipid fatty acid measurements and a protective effect of n-3 plasma phospholipid fatty acids142. Mechanistically, a high-fat diet might increase colorectal cancer risk, both directly through its effect on inflammation143, stem cell regulation144 and prostanoid metabolism145, and indirectly through its effects on the gut microbiota43.
Another indirect effect on colorectal cancer comes from the stimulatory effect of hepatic bile acid synthesis by a high-fat diet. This process results in greater quantities of bile acids escaping the enterohepatic circulation and entering the colon, in which they expand the population of microorganisms responsible for their conversion to secondary bile acids, namely those containing the bile acid deconjugating enzyme, bile acid 7α-dehydratase (primarily Clostridia species)146. Strong experimental evidence suggests that the secondary bile acids lithocholic and deoxycholic acid are carcinogenic to the colon147. In a study of18 wild-type mice, the addition of 0.2% deoxycholic acid for 8–10 months to their diet induced colonic tumours in 17 mice, and cancer in 10. Furthermore, the addition of an antioxidant prevented tumour formation, again suggesting that a balanced diet containing fat and antioxidant phytochemicals from fruits and vegetables can prevent the inflammatory effects of procarcinogens147. Evidence also indicates that bile acids might be carcinogenic to the oesophagus in Barrett oesophagus148, and to the gallbladder where chronic bacterial infection might promote the formation of secondary salts149. In the same way, substantial experimental evidence suggests that the carcinogenic potential of meat and fat-stimulated metabolites are potentiated by colonic butyrate deficiency115–119,147,150,151.
The evidence that fat directly affects gut microbiota composition and function in humans is fairly weak, and evidence that patients with steatorrhoea due to malabsorption or pancreatic insufficiency suffer an increased rate of colorectal cancer is lacking. A study in rats has shown that a change from a low-fat diet to a high-fat diet changes the microbiota composition (measured broadly by PCR) with an increase in the ratio of Firmicutes to Bacteriodetes152, but this finding could equally have been a consequence of the associated reduction of carbohydrate intake to maintain isocaloric conditions. Similarly, another study showed that a high-fat diet substantially changed the microbiota measured by terminal restriction fragment-length polymorphism in mice, with a decrease in the order Lactobacillales and an increase in the Clostridium subcluster XIVa153, but again this result could be a consequence of reduced carbohydrate intake. What is intriguing about these two studies is that in the first, supplementation of the high-fat diet with polyphenols from berries, and in the second, supplementation with a fibre source (agaro-oligosaccharides) suppressed the gut microbiota changes. Furthermore, fibre supplementation suppressed the development of AOM-generated aberrant crypt foci in the colon of mice treated with a high-fat diet, thereby inhibiting colon carcinogenesis153. These studies emphasize the fact that colorectal cancer risk is determined by dietary balance, and that the conclusions of dietary studies must take into consideration the modifying effects of luminal metabolism.
Finally, a study in a mouse model deficient in IL-10 showed that a diet high in saturated fat cultivated a blossom of Bilophila wadsworthia in the colon, which released hydrogen sulfide from taurine-containing bile acids, leading to acute inflammation that might predispose to neoplastic change75. Although hydrogen sulfide has been shown to be genotoxic in experimental cell lines76, the study in IL-10 deficient mice, in which only saturated fat and not polyunsaturated fat stimulated the growth of B. wadsworthia, suggests that all fats might not be equal.
Good dietary oils
Notably, not all fats are the same: it is not only the quantity of fat that is important, but also its composition. For example, in a study in mice154, a diet high in fish oil had no appreciable stimulatory effect on colonic 7α-dehydroxylating enzyme content, or the excretion of secondary bile acids. The balance between n-6 and n-3 fatty acid intake might be important, as exemplified by the Australian study that showed that plasma phospholipid saturated fatty acid concentrations were associated with high cancer risk, whereas high n-3 polyunsaturated fatty acid levels were associated with low risk142. Despite progressive westernization, n-3 fatty acid content in red blood cells continues to be considerably higher in Alaska Native people than in western societies, resulting in a ratio of n-6:n-3 fatty acids of approximately 1:1, compared with 10–30:1 in US white people155,156. Fish oils are a rich source of n-3 fatty acids, which have their own anti-inflammatory and antiproliferative properties157–159, in contrast to the pro-inflammatory and tumorigenic n-6 fatty acids that predominate in the western diet156. Thus, Alaska Natives should be protected from colorectal cancer by their high consumption of fish oil, and yet they have the highest reported rate in the world2,160. Interestingly, all Arctic native people are reported to have high rates of colorectal cancer160. The explanation for this finding is currently unknown, but might be related to their extremely low intakes of fibre and phytochemical-rich foods. This interpretation raises the intriguing possibility that dietary intervention in Alaska Native people aimed at increasing butyrogenesis and polyphenol intake will have profound antineoplastic effects and obliterate their excessive rate of colorectal cancer deaths. Indeed, experimental evidence shows that when provided together, butyrate and n-3 fatty acids have synergistic antineoplastic effects161,162.
Obesity
Obesity (BMI >30 kg/m2) is becoming increasingly prevalent in the USA; one survey found that more than one-third (34.9%) of adults were obese in 2011–2012 (REF 163). Obesity increases the risk of most cancers, including colorectal cancer, possibly through the generation of a chronic inflammatory state and the associated hormonal dysregulation, including hyperinsulinaemia, insulin resistance, and raised leptin and oestrogen levels, which all might contribute to a hyperproliferative state in the colonic mucosa164,165. Obesity is also commonly associated with changes in gut microbiota composition, but the reported changes are inconsistent and are probably more likely to reflect the composition of the diet166. In Gordon’s studies on distal gut microbiota of genetically obese mice and their lean littermates, as well as those of obese and lean human volunteers, they showed that the microbiome associated with obesity had an increased capacity to harvest energy from the diet167. The obese microbiome was enriched with functional groups involved in starch degradation and butyrate production, and faecal content of fermentation products was increased. Remarkably, when they transplanted the microbiome from obese mice to germfree mice they found a significantly (P <0.05) greater increase in total body fat than colonization with the microbiota from lean mice despite isocaloric feeding. However, others have reported different changes in the ratios of Firmicutes to Bacteriodes in humans with obesity168 and the dream of being able to change the risk of obesity by changing the gut microbiota has not yet been realized: the situation is clearly far more complex as reviewed elsewhere168,169.
Evidence suggests that the high-fat content of the obesogenic diet drives carcinogenesis. For example, in a study comparing African Americans and rural South Africans, obesity in South Africans was associated with high intakes of carbohydrate and not fat, in tandem with low colonic bile acids, low biomarkers of colon cancer risk, and low colon cancer rates in the population170. Furthermore, a study in a mouse model genetically susceptible to intestinal tumorigenesis demonstrated that a high-fat diet promoted tumour progression independently of obesity43.
How can colon cancer be prevented?
Fibre supplementation
Owing to the strong experimental evidence that butyrate suppresses colorectal neoplasia, multiple clinical trials have been conducted to determine whether fibre supplementation could reduce the risk of colorectal polyp recurrences. Unfortunately, few studies were positive171, most likely because the level of fibre supplementation achieved was insufficient. For example, the most-quoted Polyp Prevention Trial, in which 2,079 participants with a previous history of colorectal polyps were randomly assigned to an advised low-fat, high-fibre diet, found no overall difference in polyp recurrence after 4 years and 8 years compared with a control group given a brochure on healthy eating and assigned to follow their usual diet172. However, the high-fibre group were estimated to have consumed an average of only 32 g per day, which might have been further reduced as compliance was low, indicated by the change in biomarkers of fat and green vegetable intake (blood cholesterol and vitamin A, respectively).
What is the threshold intake of fibre?
We now have strong human and experimental evidence that there is a threshold level for fibre consumption, above which the level of butyrogenesis is sufficient to overcome the inflammatory, neoplastic and carcinogenic potential of nitrogenous microbial metabolites (such as ammonia and nitrosamines) and environmental carcinogens (such as polycyclic aromatic hydrocarbons and heterocyclic amines). First, the traditional African diet identified to be protective from colon cancer contained >50 g fibre per day173. Second, increasing the fibre consumption in African Americans at high risk of colorectal cancer to 55 g per day was associated with a substantial decrease in colonic mucosal biomarkers of cancer risk (epithelial proliferation) within 2 weeks of change (discussed in the next section)50. Third, in the Polyp Prevention Trial, a significantly reduced odds ratio for advanced (>1 cm, >25% villous, or high-grade dysplasia) adenomatous polyp recurrence was found in the subgroup consuming the highest quartile of high fibre beans174. These observations suggest that the required fibre consumption level should be >50 g per day and that the recommended intake level in the USA of 25 g per day for women and 38 g per day for men, which was based on the level required to maintain cardiovascular health, needs revising175,176. Fibre recommendations are similar in the UK, Europe, Australia and New Zealand177–179.
Changing diet to change the microbiota
A series of studies have been performed over the past 20 years to identify the dramatic difference in colorectal cancer prevalence between African Americans (65 cases per 100,000) and native Africans (<5 cases per 100,000)49–51,180–186. One study provided evidence that the fibre and fat content of the diet were chiefly responsible, and that the effect of these diets on biomarkers of cancer risk were mediated by the microbial metabolites50. This study was unique in that it was tightly controlled and what the participants consumed and returned was examined under observation for the total duration of the 2-week in-house dietary intervention50. Diets were switched such that the healthy middle-aged African Americans increased their fibre intake to 55 g per day and reduced their fat intake to 51 g per day, while the rural Africans reduced their fibre intake to 7 g per day and increased fat intake to 134 g per day. Remarkably, epithelial proliferative rates in colonic mucosal biopsies taken from African Americans decreased to levels lower than those of rural Africans at baseline, whereas rates increased in rural Africans to levels greater than the African Americans at baseline50. Inflammatory biomarkers followed a similar pattern, with a decrease in CD3+ intraepithelial lymphocytes and CD68+ lamina propria macrophages in African Americans and an increase in rural Africans. These changes were accompanied by substantial reciprocal changes in the gut microbiota and metabolome, and specifically in microbial butyrate and secondary bile acid production, supporting the hypothesis that mucosal biomarkers of cancer risk are modified by the diet through the microbiota. Reciprocal changes in urinary metabolites derived from microbial metabolism of green vegetables were also observed following the diet switch, suggesting that some of the reduction in mucosal proliferation in African Americans could also have been related to the effects of increased phytochemical consumption in the Africanized diets.
A further fascinating finding was that the mucosal biopsy samples from all the rural Africans at baseline showed intense lymphocytic infiltration consistent with lymphocytic colitis, (associated with parasitic infections in some) despite the absence of any gastrointestinal symptoms50. That this finding has not led to an increase in colon cancer within the community is surprising considering the strong link between chronic inflammation and neoplasia187. One can speculate that the high rate of butyrogenesis arrests neoplastic progression which, if true, implies that the inevitable progressive global westernization and associated reduced consumption of fibre-rich foods will lead to an explosion in colorectal cancer incidence.
Conclusions
Evidence based on controlled epidemiological, cell culture, animal and human studies has been presented to support the view that the high incidence of colorectal cancer in westernized countries is a consequence of dietary imbalance, principally a deficiency of high-fibre fruits, vegetables and whole grains. Evidence has been presented that the evolution of man has been accompanied by high complex carbohydrate feeding patterns, which have a profound effect on the colonic microbiota and their ability to produce metabolites that support colonic health and prevent carcinogenesis. Industrialization, westernization and food refinement has resulted in disturbance of the homeostatic role of our gut microbiota due to ‘starvation’ of the microbiota, impaired butyrogenesis and loss of host defense against environmental carcinogens. National guidelines for fibre requirements have not been based on robust scientific studies. Such studies are urgently needed to define the ideal metabolic requirements of the gut microbiota, which, when met, could result in a suppression of colorectal cancer risk in westernized communities to that of rural Africa.
Key points.
Colorectal cancer is a so-called westernized disease and the second leading cause of cancer death worldwide; approximately half of those with the disease will die from it
Geographical variation, migration studies and experimental studies provide compelling evidence that environmental factors, rather than genetic dysfunction, are responsible for the development of colorectal cancer
Convincing evidence suggests that risk of colon cancer is increased by processed and unprocessed meat consumption but suppressed by fibre-rich foods
Dietary risk is mediated by the colonic microbiota; carbohydrate residues stimulate production of metabolites that maintain mucosal health, proteinaceous residues and fat-stimulated bile acids might result in pro-inflammatory and carcinogenic metabolites
A moderate intake of meat and fat is part of our omnivorous diet and the carcinogenic potential can be suppressed by the induction of butyrogenesis from fibre-rich foods
Current dietary fibre recommendations need to be reviewed as they are based on the maintenance of cardiovascular health and are below the levels associated with low colon cancer risk
Footnotes
Competing interests statement
The author declares no competing interests.
References
- 1.Ferlay J et al. GLOBOCAN 2012 v1.0, Cancer incidence and mortality worldwide: IARC CancerBase No. 11 [online]. Lyon, France: International Agency for Research on Cancer, http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx?cancer=colorectal (2013). [Google Scholar]
- 2.Perdue DG, Haverkamp D, Perkins C, Daley CM & Provost E Geographic variation in colorectal cancer incidence and mortality, age of onset, and stage at diagnosis among American Indian and Alaska Native people, 1990–2009. Am. J. Public Health 104, S404–S414 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Houlston RS et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat. Genet. 42, 973–977 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Figueiredo JC et al. Genome-wide diet–gene interaction analyses for risk of colorectal cancer. PLoS Genet. 10, e1004228 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aune D et al. Dietary fibre, whole grains, and risk of colorectal cancer: systematic review and dose-response meta-analysis of prospective studies. BMJ 343, d6617 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Magalhães B, Peleteiro B & Lunet N Dietary patterns and colorectal cancer: systematic review and meta-analysis. Eur. J. Cancer Prev. 21, 15–23 (2012). [DOI] [PubMed] [Google Scholar]
- 7.World Cancer Research Fund & American Institute for Cancer Research. Colorectal cancer 2011 report. Food, nutrition, physical activity, and the prevention of colorectal cancer. WCRF; http://www.wcrf.org/sites/default/files/Colorectal-Cancer-2011-Report.pdf (2011). [Google Scholar]
- 8.World Health Organization. IARC monographs evaluate consumption of red meat and processed meat. ¡ARC; https://www.iarc.fr/en/media-centre/pr/2015/pdfs/pr240_E.pdf (2015). [Google Scholar]
- 9.Doll R & Peto R The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J. Natl Cancer ¡nst. 66, 1191–1308 (1981). [PubMed] [Google Scholar]
- 10.Blot WJ & Tarone RE Doll and Peto’s quantitative estimates of cancer risks: holding generally true for 35 years. J. Natl Cancer ¡nst. 107, djv044 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Le Marchand L & Kolonel LN Cancer in Japanese migrants to Hawaii: interaction between genes and environment [French]. Rev. Épidémiol. Santé Publique 40, 425–430 (1992). [PubMed] [Google Scholar]
- 12.Nosho K et al. Association of Fusobacterium nucleatum with immunity and molecular alterations in colorectal cancer. World J. Gastroenterol. 22, 557–566 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tjalsma H, Boleij A, Marchesi JR & Dutilh BE A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat. Rev. Microbiol. 10, 575–582 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Colnot S et al. Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Lab. Invest. 84 1619–1630 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Bordonaro M, Lazarova DL & Sartorelli AC Butyrate and Wnt signaling: a possible solution to the puzzle of dietary fiber and colon cancer risk? Cell Cycle 7, 1178–1183 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Beyer-Sehlmeyera G et al. Butyrate is only one of several growth inhibitors produced during gut flora-mediated fermentation of dietary fibre sources. Br. J. Nutr. 90, 1057–1070 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Bultman SJ Molecular pathways: geneenvironment interactions regulating dietary fiber induction of proliferation and apoptosis via butyrate for cancer prevention. Clin. Cancer Res. 20, 799–803 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verma M Cancer control and prevention: nutrition and epigenetics. Curr. Opin. Clin. Nutr. Metab. Care 16, 376–384 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Tarapore RS, Siddiqui IA & Mukhtar H Modulation of Wnt/β-catenin signaling pathway by bioactive food components. Carcinogenesis 33, 483–491 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fung KY, Cosgrove L, Lockett T, Head R & Topping DL A review of the potential mechanisms for the lowering of colorectal oncogenesis by butyrate. Br. J. Nutr. 108, 820–831 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Matsuki T et al. Epithelial cell proliferation arrest induced by lactate and acetate from Lactobacillus casei and Bifidobacterium breve. PLoS ONE 8, e63053 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu Y et al. Manipulation of the gut microbiota using resistant starch is associated with protection against colitis-associated colorectal cancer in rats. Carcinogenesis 37, 366–375 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Cho M, Carter J, Harari S & Pei Z The interrelationships of the gut microbiome and inflammation in colorectal carcinogenesis.Clin. Lab. Med. 34, 699–710 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greer JB & O’Keefe SJ Microbial induction of immunity, inflammation and cancer. Front. Physiol. 1, 168 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vipperla K & O’Keefe SJ Diet, microbiota, and dysbiosis: a ‘recipe’ for colorectal cancer. Food Funct. 7, 1731–1740 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berg DJ et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4 + TH1 -like responses. J. Clin. Invest. 98, 1010–1020 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uronis JM et al. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS ONE 4, e6026 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borges-Canha M, Portela-Cidade JP, Dinis Ribeiro M, Leite-Moreira AF & Pimentel-Nunes P Role of colonic microbiota in colorectal carcinogenesis: a systematic review. Rev. Esp. Enferm. Dig. 107, 659–671 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Baxter NT, Zackular JP, Chen GY & Schloss PD Structure of the gut microbiome following colonization with human feces determines colonic tumor burden. Microbiome 2, 20 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zackular JP et al. The gut microbiome modulates colon tumorigenesis. mBio 4, e00692–13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeller G et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol.10, 766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kostic AD et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14, 207–215 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mima K et al. Fusobacterium nucleatum and T cells in colorectal carcinoma. JAMA Oncol. 1, 653–661 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tahara T et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 74, 1311–1318 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu S et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15, 1016–1022 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toprak NU et al. A possible role of Bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin. Microbiol. Infect. 12, 782–786 (2006). [DOI] [PubMed] [Google Scholar]
- 37.DeStefano Shields CE et al. Reduction of murine colon tumorigenesis driven by enterotoxigenic Bacteroides fragilis using cefoxitin treatment. J. Infect. Dis. 214, 122–129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Urbaniak C et al. Microbiota of human breast tissue. Appl. Environ. Microbiol. 80, 3007–3014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Al-Asmakh M et al. The gut microbiota and developmental programming of the testis in mice. PLoS ONE 9, e103809 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collado MC, Rautava S, Aakko J, Isolauri E & Salminen S Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci. Rep. 6, 23129(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheflin AM, Whitney AK & Weir TL Cancer-promoting effects of microbial dysbiosis. Curr. Oncol. Rep. 16, 406 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xuan C et al. Microbial dysbiosis is associated with human breast cancer. PLoS ONE 9, e83744 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schulz MD et al. High-fat-diet-mediated dysbiosis promotes intestinal carcinogenesis independently of obesity. Nature 514, 508–512 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rook GA 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: Darwinian medicine and the ‘hygiene’ or ‘old friends’ hypothesis. Clin. Exp. Immunol. 160, 70–79 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cordain L et al. Origins and evolution of the Western diet: health implications for the 21st century. Am. J. Clin. Nutr. 81, 341–354 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Balter V, Braga J, Telouk P & Thackeray JF Evidence for dietary change but not landscape use in South African early hominins. Nature 489, 558–560 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Richardson EU Archaeological dig reveals causes — and possible cures — for diabetes epidemic. Indian Country Today Media Network [online] http://indiancountrytodaymedianetwork.com/2012/08/23/archaeological-dig-reveals-causes-and-possible-cures-diabetes-epidemic-130651 (2012) [Google Scholar]
- 48.Ungar PS & Sponheimer M The diets of early hominins. Science 334, 190–193 (2011). [DOI] [PubMed] [Google Scholar]
- 49.O’Keefe SJ et al. Why do African Americans get more colon cancer than Native Africans? J. Nutr. 137, 175S–182S (2007). [DOI] [PubMed] [Google Scholar]
- 50.O’Keefe SJ et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 6, 6342 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ou J et al. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am. J. Clin. Nutr. 98, 111–120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Macfarlane GT & Englyst HN Starch utilization by the human large intestinal microflora. J. Appl. Bacteriol. 60, 195–201 (1986). [DOI] [PubMed] [Google Scholar]
- 53.Macfarlane GT, Gibson GR & Cummings JH Comparison of fermentation reactions in different regions of the human colon. J. Appl. Bacteriol. 72, 57–64 (1992). [DOI] [PubMed] [Google Scholar]
- 54.Cummings JH, Beatty ER, Kingman SM, Bingham SA & Englyst HN Digestion and physiological properties of resistant starch in the human large bowel. Br. J. Nutr. 75, 733–747 (1996). [DOI] [PubMed] [Google Scholar]
- 55.Roediger WE Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology 83, 424–429 (1982). [PubMed] [Google Scholar]
- 56.Flint HJ, Duncan SH, Scott KP & Louis P Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 74, 13–22 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Macfarlane S & Macfarlane GT Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 62, 67–72 (2003). [DOI] [PubMed] [Google Scholar]
- 58.Macfarlane GT & Macfarlane S Bacteria, colonic fermentation, and gastrointestinal health. J. AOAC Int. 95,50–60 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Duncan SH et al. Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl. Environ. Microbiol. 73, 1073–1078 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Waldecker M, Kautenburger T, Daumann H, Busch C & Schrenk D Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 19, 587–593 (2008). [DOI] [PubMed] [Google Scholar]
- 61.Macfarlane GT & Macfarlane S Fermentation in the human large intestine: its physiologic consequences and the potential contribution of prebiotics. J. Clin. Gastroenterol. 45, S120–S127 (2011). [DOI] [PubMed] [Google Scholar]
- 62.Frost G et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 5, 3611 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Al-Lahham SH, Peppelenbosch MP, Roelofsen H, Vonk RJ & Venema K Biological effects of propionic acid in humans; metabolism, potential applications and underlying mechanisms. Biochim. Biophys. Acta 1801, 1175–1183 (2010). [DOI] [PubMed] [Google Scholar]
- 64.Venter CS, Vorster HH & Cummings JH Effects of dietary propionate on carbohydrate and lipid metabolism in healthy volunteers. Am. J. Gastroenterol. 85, 549–553 (1990). [PubMed] [Google Scholar]
- 65.Canfora EE, Jocken JW & Blaak EE Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 11, 577–591 (2015). [DOI] [PubMed] [Google Scholar]
- 66.Nordgaard I, Hansen BS & Mortensen PB Importance of colonic support for energy absorption as small-bowel failure proceeds. Am. J. Clin. Nutr. 64, 222–231 (1996). [DOI] [PubMed] [Google Scholar]
- 67.Silvester KR & Cummings JH Does digestibility of meat protein help explain large bowel cancer risk? Nutr. Cancer 24, 279–288 (1995). [DOI] [PubMed] [Google Scholar]
- 68.Windey K, De Preter V & Verbeke K Relevance of protein fermentation to gut health. Mol. Nutr. Food Res. 56, 184–196 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Bui TP et al. Production of butyrate from lysine and the Amadori product fructoselysine by a human gut commensal. Nat. Commun. 6, 10062 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Visek WJ Diet and cell growth modulation by ammonia. Am. J. Clin. Nutr. 31, S216–S220 (1978). [DOI] [PubMed] [Google Scholar]
- 71.Clinton SK, Bostwick DG, Olson LM, Mangian HJ & Visek WJ Effects of ammonium acetate and sodium cholate on N-methyl-N’-nitro-N-nitrosoguanidine-induced colon carcinogenesis of rats. Cancer Res. 48, 3035–3039 (1988). [PubMed] [Google Scholar]
- 72.Mirvish SS Role of N-nitroso compounds (NOC) and N-nitrosation in etiology of gastric, esophageal, nasopharyngeal and bladder cancer and contribution to cancer of known exposures to NOC. Cancer Lett. 93,17–48 (1995). [DOI] [PubMed] [Google Scholar]
- 73.Cross AJ & Sinha R Meat-related mutagens/ carcinogens in the etiology of colorectal cancer. Environ. Mol. Mutagen. 44, 44–55 (2004). [DOI] [PubMed] [Google Scholar]
- 74.Bingham SA et al. Does increased endogenous formation of N-nitroso compounds in the human colon explain the association between red meat and colon cancer? Carcinogenesis 17, 515–523 (1996). [DOI] [PubMed] [Google Scholar]
- 75.Devkota S et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/−mice. Nature. 487, 104–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Attene-Ramos MS, Wagner ED, Plewa MJ & Gaskins HR Evidence that hydrogen sulfide is a genotoxic agent. Mol. Cancer Res. 4, 9–14 (2006). [DOI] [PubMed] [Google Scholar]
- 77.Wallace JL, Vong L, McKnight W, Dicay M & Martin GR Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology 137, 569–578, (2009). [DOI] [PubMed] [Google Scholar]
- 78.Ianaro A, Cirino G & Wallace JL Hydrogen sulfide-releasing anti-inflammatory drugs for chemoprevention and treatment of cancer. Pharmacol. Res. 111, 652–658 (2016). [DOI] [PubMed] [Google Scholar]
- 79.Elsheikh W, Blackler RW, Flannigan KL & Wallace JL Enhanced chemopreventive effects of a hydrogen sulfide-releasing anti-inflammatory drug (ATB-346) in experimental colorectal cancer. Nitric Oxide 41, 131–137 (2014). [DOI] [PubMed] [Google Scholar]
- 80.De Preter V et al. Decreased mucosal sulfide detoxification is related to an impaired butyrate oxidation in ulcerative colitis. Inflamm. Bowel Dis. 18, 2371–2380 (2012). [DOI] [PubMed] [Google Scholar]
- 81.McCall IC et al. Effects of phenol on barrier function of a human intestinal epithelial cell line correlate with altered tight junction protein localization. Toxicol. Appl. Pharmacol. 241, 61–70 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Birkett A, Muir J, Phillips J, Jones G & O’Dea K Resistant starch lowers fecal concentrations of ammonia and phenols in humans. Am. J. Clin. Nutr. 63, 766–772 (1996). [DOI] [PubMed] [Google Scholar]
- 83.De Preter V et al. The in vivo use of the stable isotope-labelled biomarkers lactose-[l5N]ureide and [2H4]tyrosine to assess the effects of pro- and prebiotics on the intestinal flora of healthy human volunteers. Br. J. Nutr. 92, 439–446 (2004). [DOI] [PubMed] [Google Scholar]
- 84.Rafter J et al. Dietary synbiotics reduce cancer risk factors in polypectomized and colon cancer patients. Am. J. Clin. Nutr. 85, 488–496 (2007). [DOI] [PubMed] [Google Scholar]
- 85.Humphreys KJ et al. Dietary manipulation of oncogenic microRNA expression in human rectal mucosa: a randomized trial. Cancer Prev. Res. (Phila.) 7, 786–795 (2014). [DOI] [PubMed] [Google Scholar]
- 86.Hu S, Liu L, Chang EB, Wang JY & Raufman JP Butyrate inhibits pro-proliferative miR-92a by diminishing c-Myc-induced miR-17–92a cluster transcription in human colon cancer cells. Mol. Cancer 14, 180 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen HM et al. Decreased dietary fiber intake and structural alteration of gut microbiota in patients with advanced colorectal adenoma. Am. J. Clin. Nutr. 97, 1044–1052 (2013). [DOI] [PubMed] [Google Scholar]
- 88.Wang T et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 6, 320–329 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vipperla K & O’Keefe SJ The microbiota and its metabolites in colonic mucosal health and cancer risk. Nutr. Clin. Pract. 27, 624–635 (2012). [DOI] [PubMed] [Google Scholar]
- 90.Rodriguez-Cabezas ME et al. Dietary fiber down-regulates colonic tumor necrosis factor a and nitric oxide production in trinitrobenzenesulfonic acid-induced colitic rats. J. Nutr. 132, 3263–3271 (2002). [DOI] [PubMed] [Google Scholar]
- 91.Inan MS et al. The luminal short-chain fatty acid butyrate modulates NF-kB activity in a human colonic epithelial cell line. Gastroenterology 118, 724–734 (2000). [DOI] [PubMed] [Google Scholar]
- 92.Chirakkal H et al. Upregulation of BAK by butyrate in the colon is associated with increased Sp3 binding. Oncogene 25, 7192–7200 (2006). [DOI] [PubMed] [Google Scholar]
- 93.Hinnebusch BF, Meng S, Wu JT, Archer SY & Hodin RA The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J. Nutr. 132, 1012–1017 (2002). [DOI] [PubMed] [Google Scholar]
- 94.Comalada M et al. The effects of short-chain fatty acids on colon epithelial proliferation and survival depend on the cellular phenotype. J. Cancer Res. Clin. Oncol. 132, 487–497 (2006). [DOI] [PubMed] [Google Scholar]
- 95.Andoh A, Shimada M, Araki Y, Fujiyama Y & Bamba T Sodium butyrate enhances complement-mediated cell injury via down-regulation of decay-accelerating factor expression in colonic cancer cells. Cancer Immunol. Immunother. 50, 663–672 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rodriguez-Salvador J et al. Effect of sodium butyrate on pro-matrix metalloproteinase-9 and −2 differential secretion in pediatric tumors and cell lines. J. Exp. Clin. Cancer Res. 24, 463–473 (2005). [PubMed] [Google Scholar]
- 97.Zeng H & Briske-Anderson M Prolonged butyrate treatment inhibits the migration and invasion potential of HT1080 tumor cells. J. Nutr. 135, 291–295 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Zgouras D, Wachtershauser A, Frings D & Stein J Butyrate impairs intestinal tumor cell-induced angiogenesis by inhibiting HIF-1a nuclear translocation. Biochem. Biophys. Res. Commun. 300, 832–838 (2003). [DOI] [PubMed] [Google Scholar]
- 99.Willemsen LE, Koetsier MA, van Deventer SJ & van Tol EA Short chain fatty acids stimulate epithelial mucin 2 expression through differential effects on prostaglandin E1 and E2 production by intestinal myofibroblasts. Gut 52, 1442–1447 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.D’Argenio G et al. Butyrate enemas in experimental colitis and protection against large bowel cancer in a rat model. Gastroenterology 110, 1727–1734 (1996). [DOI] [PubMed] [Google Scholar]
- 101.Schauber J et al. Expression of the cathelicidin LL-37 is modulated by short chain fatty acids in colonocytes: relevance of signalling pathways. Gut 52, 735–741 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Malago JJ, Koninkx JF, Tooten PC, van Liere EA & van Dijk JE Anti-inflammatory properties of heat shock protein 70 and butyrate on Salmonella-induced interleukin-8 secretion in enterocyte-like Caco-2 cells. Clin. Exp. Immunol. 141, 62–71 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Smith PM et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hu S et al. The microbe-derived short chain fatty acid butyrate targets miRNA-dependent p21 gene expression in human colon cancer. PLoS ONE 6, e16221 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.He L et al. A microRNA polycistron as a potential human oncogene. Nature 435, 828–833 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li C-J, Li RW & Elsasser TH MicroRNA (miRNA) expression is regulated by butyrate-induced epigenetic modulation of gene expression in bovine cells. Genet. Epigenet. 3, 23–32 (2010). [Google Scholar]
- 107.Humphreys KJ, Cobiac L, Le Leu RK, Van der Hoek MB & Michael MZ Histone deacetylase inhibition in colorectal cancer cells reveals competing roles for members of the oncogenic miR-17–92 cluster. Mol. Carcinog. 52, 459–474 (2013). [DOI] [PubMed] [Google Scholar]
- 108.Sengupta S, Muir JG & Gibson PR Does butyrate protect from colorectal cancer? J. Gastroenterol. Hepatol. 21, 209–218 (2006). [DOI] [PubMed] [Google Scholar]
- 109.Donohoe DR et al. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 48, 612–626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Donohoe DR et al. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 4, 1387–1397 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Belcheva A et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 158, 288–299 (2014). [DOI] [PubMed] [Google Scholar]
- 112.Ye H, Liu J, Feng P, Zhu W & Mao S Grain-rich diets altered the colonic fermentation and mucosa-associated bacterial communities and induced mucosal injuries in goats. Sci. Rep. 6, 20329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gressley TF, Hall MB & Armentano LE Ruminant Nutrition Symposium: productivity, digestion, and health responses to hindgut acidosis in ruminants. J. Anim. Sci. 89, 1120–1130 (2011). [DOI] [PubMed] [Google Scholar]
- 114.Kowlgi NG & Chhabra L D-lactic acidosis: an underrecognized complication of short bowel syndrome. Gastroenterol. Res. Pract. 2015, 476215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Winter J et al. Inhibition by resistant starch of red meat-induced promutagenic adducts in mouse colon. CancerPrev. Res. (Phila.) 4, 1920–1928 (2011). [DOI] [PubMed] [Google Scholar]
- 116.Toden S, Bird AR, Topping DL & Conlon MA Resistant starch prevents colonic DNA damage induced by high dietary cooked red meat or casein in rats. Cancer Biol. Ther. 5, 267–272 (2006). [DOI] [PubMed] [Google Scholar]
- 117.Toden S, Bird AR, Topping DL & Conlon MA Differential effects of dietary whey, casein and soya on colonic DNA damage and large bowel SCFA in rats fed diets low and high in resistant starch. Br. J. Nutr. 97535–543 (2007). [DOI] [PubMed] [Google Scholar]
- 118.Hylla S et al. Effects of resistant starch on the colon in healthy volunteers: possible implications for cancer prevention. Am. J. Clin. Nutr. 67, 136–142 (1998). [DOI] [PubMed] [Google Scholar]
- 119.van Munster IP, Tangerman A & Nagengast FM Effect of resistant starch on colonic fermentation, bile acid metabolism, and mucosal proliferation. Dig. Dis. Sci. 39, 834–842 (1994). [DOI] [PubMed] [Google Scholar]
- 120.Scharlau D et al. Mechanisms of primary cancer prevention by butyrate and other products formed during gut flora-mediated fermentation of dietary fibre. Mutat. Res. 682, 39–53 (2009). [DOI] [PubMed] [Google Scholar]
- 121.Cardona F, Andres-Lacueva C, Tulipani S, Tinahones FJ & Queipo-Ortuno MI Benefits of polyphenols on gut microbiota and implications in human health. J. Nutr. Biochem. 24, 1415–1422 (2013). [DOI] [PubMed] [Google Scholar]
- 122.Russell WR et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am. J. Clin. Nutr. 93, 1062–1072 (2011). [DOI] [PubMed] [Google Scholar]
- 123.Dueñas M et al. A survey of modulation of gut microbiota by dietary polyphenols. Biomed. Res. Int. 2015, 850902 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kemperman RA, Bolca S, Roger LC & Vaughan EE Novel approaches for analysing gut microbes and dietary polyphenols: challenges and opportunities. Microbiology 156, 3224–3231 (2010). [DOI] [PubMed] [Google Scholar]
- 125.Sirk TW, Friedman M & Brown EF Molecular binding of black tea theaflavins to biological membranes: relationship to bioactivities. J. Agric. Food Chem. 59, 3780–3787 (2011). [DOI] [PubMed] [Google Scholar]
- 126.Martin C, Zhang Y, Tonelli C & Petroni K Plants, diet, and health. Annu. Rev. Plant Biol. 64, 19–46 (2013). [DOI] [PubMed] [Google Scholar]
- 127.Eid N et al. Impact of palm date consumption on microbiota growth and large intestinal health: a randomised, controlled, cross-over, human intervention study. Br. J. Nutr. 114, 1226–1236 (2015). [DOI] [PubMed] [Google Scholar]
- 128.Nuñez-Sánchez MA et al. In vivo relevant mixed urolithins and ellagic acid inhibit phenotypic and molecular colon cancer stem cell features: a new potentiality for ellagitannin metabolites against cancer. Food Chem. Toxicol. 92, 8–16 (2016). [DOI] [PubMed] [Google Scholar]
- 129.Giménez-Bastida JA et al. Intestinal ellagitannin metabolites ameliorate cytokine-induced inflammation and associated molecular markers in human colon fibroblasts. J. Agric. Food Chem. 60, 8866–8876 (2012). [DOI] [PubMed] [Google Scholar]
- 130.González-Sarrías A, Nuñez-Sánchez MA, García-Villalba R, Tomás-Barberán FA & Espín JC Antiproliferative activity of the ellagic acid-derived gut microbiota isourolithin A and comparison with its urolithin A isomer: the role of cell metabolism. Eur. J. Nutr. 10.1007/s00394-015-1131-7 (2015). [DOI] [PubMed] [Google Scholar]
- 131.Wang LS et al. A phase Ib study of the effects of black raspberries on rectal polyps in patients with familial adenomatous polyposis. Cancer Prev. Res. (Phila.) 7, 666–674 (2014). [DOI] [PubMed] [Google Scholar]
- 132.Stoner GD et al. Cancer prevention with freeze-dried berries and berry components. Semin. Cancer Biol. 17, 403–410 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Queipo-Ortuno MI et al. Influence of red wine polyphenols and ethanol on the gut microbiota ecology and biochemical biomarkers. Am. J. Clin. Nutr.95, 1323–1334 (2012). [DOI] [PubMed] [Google Scholar]
- 134.Yamada H et al. Coffee consumption and risk of colorectal cancer: the Japan Collaborative Cohort Study. J. Epidemiol. 24, 370–378 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Guercio BJ et al. Coffee intake, recurrence, and mortality in stage III colon cancer: results from CALGB 89803 (Alliance). J. Clin. Oncol. 33, 3598–3607 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang ZJ et al. Dietary polyphenols and colorectal cancer risk: the Fukuoka colorectal cancer study. World J. Gastroenterol. 19, 2683–2690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schmit SL, Rennert HS, Rennert G & Gruber SB Coffee consumption and the risk of colorectal cancer. Cancer Epidemiol. Biomarkers Prev. 25, 634–639 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Guertin KA et al. Serum biomarkers of habitual coffee consumption may provide insight into the mechanism underlying the association between coffee consumption and colorectal cancer. Am. J. Clin. Nutr. 1011000–1011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Willett WC, Stampfer MJ, Colditz GA, Rosner BA & Speizer FE Relation of meat, fat, and fiber intake to the risk of colon cancer in a prospective study among women. N. Engl. J. Med. 323, 1664–1672 (1990). [DOI] [PubMed] [Google Scholar]
- 140.Kato I, Majumdar AP, Land SJ, Barnholtz-Sloan JS & Severson RK Dietary fatty acids, luminal modifiers, and risk of colorectal cancer. Int. J. Cancer 127, 942–951 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Enos RT et al. High-fat diets rich in dietary saturated fat protect against azoxymethane/dextran sulfate sodium induced colon cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 310, G906–G919 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hodge AM et al. Dietary and biomarker estimates of fatty acids and risk of colorectal cancer. Int. J. Cancer 137, 1224–1234 (2015). [DOI] [PubMed] [Google Scholar]
- 143.Viggiano E et al. Effects of an high-fat diet enriched in lard or in fish oil on the hypothalamic amp-activated protein kinase and inflammatory mediators. Front. Cell. Neurosci. 10, 150 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Beyaz S et al. High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531, 53–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang D & Dubois RN Eicosanoids and cancer. Nat. Rev. Cancer 10, 181–193 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wells JE & Hylemon PB Identification and characterization of a bile acid 7α-dehydroxylation operon in Clostridium sp. strain TO-931, a highly active 7α-dehydroxylating strain isolated from human feces. Appl. Environ. Microbiol. 66, 1107–1113 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bernstein C et al. Carcinogenicity of deoxycholate, a secondary bile acid. Arch. Toxicol. 85, 863–871 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Burnat G, Majka J & Konturek PC Bile acids are multifunctional modulators of the Barrett’s carcinogenesis. J. Physiol. Pharmacol. 61, 185–192 (2010). [PubMed] [Google Scholar]
- 149.Sharma V, Chauhan VS, Nath G, Kumar A & Shukla VK Role of bile bacteria in gallbladder carcinoma. Hepatogastroenterology 54, 1622–1625 (2007). [PubMed] [Google Scholar]
- 150.Alberts DS et al. Randomized, double-blinded, placebo-controlled study of effect of wheat bran fiber and calcium on fecal bile acids in patients with resected adenomatous colon polyps. J. Natl Cancer Inst. 88, 81–92 (1996). [DOI] [PubMed] [Google Scholar]
- 151.De Boever P et al. Protective effect of the bile salt hydrolase-active Lactobacillus reuteri against bile salt cytotoxicity. Appl. Microbiol. Biotechnol. 53, 709–714 (2000). [DOI] [PubMed] [Google Scholar]
- 152.Taira T et al. Dietary polyphenols increase fecal mucin and immunoglobulin A and ameliorate the disturbance in gut microbiota caused by a high fat diet. J. Clin. Biochem. Nutr. 57, 212–216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Higashimura Y et al. Protective effect of agaro-oligosaccharides on gut dysbiosis and colon tumorigenesis in high-fat diet-fed mice. Am. J. Physiol. Gastrointest. Liver Physiol. 310, G367–G375 (2016). [DOI] [PubMed] [Google Scholar]
- 154.Reddy BS, Simi B, Patel N, Aliaga C & Rao CV Effect of amount and types of dietary fat on intestinal bacterial 7 α-dehydroxylase and phosphatidylinositol-specific phospholipase C and colonic mucosal diacylglycerol kinase and PKC activities during stages of colon tumor promotion. Cancer Res. 56, 2314–2320 (1996). [PubMed] [Google Scholar]
- 155.Bersamin A, Luick BR, King IB, Stern JS & Zidenberg-Cherr S Westernizing diets influence fat intake, red blood cell fatty acid composition, and health in remote Alaskan Native communities in the Center for Alaska Native Health Study. J. Am. Diet. Assoc. 108, 266–273 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Simopoulos AP Evolutionary aspects of diet, the omega-6/omega-3 ratio and genetic variation: nutritional implications for chronic diseases. Biomed. Pharmacother. 60, 502–507 (2006). [DOI] [PubMed] [Google Scholar]
- 157.Kim S, Sandler DP, Galanko J, Martin C & Sandler RS Intake of polyunsaturated fatty acids and distal large bowel cancer risk in whites and African Americans. Am. J. Epidemiol. 171, 969–979 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Woodworth HL et al. Dietary fish oil alters T lymphocyte cell populations and exacerbates disease in a mouse model of inflammatory colitis. Cancer Res. 70 7960–7969 (2010). [DOI] [PubMed] [Google Scholar]
- 159.Moreira APB et al. Fish oil ingestion reduces the number of aberrant crypt foci and adenoma in 1,2-dimethylhydrazine-induced colon cancer in rats. Braz. J. Med. Biol. Res. 42, 1167–1172 (2009). [DOI] [PubMed] [Google Scholar]
- 160.Young TK, Kelly JJ, Friborg J, Soininen L & Wong KO Cancer among circumpolar populations: an emerging public health concern. Int. J. Circumpolar Health 75, 29787 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hofmanova J, Vaculova A, Lojek A & Kozubik A Interaction of polyunsaturated fatty acids and sodium butyrate during apoptosis in HT-29 human colon adenocarcinoma cells. Eur. J. Nutr. 44, 40–51 (2005). [DOI] [PubMed] [Google Scholar]
- 162.Kolar SSN et al. Synergy between docosahexaenoic acid and butyrate elicits p53-independent apoptosis via mitochondrial Ca2+ accumulation in colonocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 293, G935–G943 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ogden CL, Carroll MD, Kit BK & Flegal KM Prevalence of obesity among adults: United States, 2011–2012. NCHS Data Brief; https://www.cdc.gov/nchs/data/databriefs/db131.pdf (2013). [PubMed] [Google Scholar]
- 164.Pischon T, Nöthlings U & Boeing H Obesity and cancer. Proc. Nutr. Soc. 67, 128–145 (2008). [DOI] [PubMed] [Google Scholar]
- 165.Goday A et al. Obesity as a risk factor in cancer: a national consensus of the Spanish Society for the Study of Obesity and the Spanish Society of Medical Oncology. Clin. Transl Oncol. 17, 763–771 (2015). [DOI] [PubMed] [Google Scholar]
- 166.Bell DS Changes seen in gut bacteria content and distribution with obesity: causation or association? Postgrad. Med. 127, 863–868 (2015). [DOI] [PubMed] [Google Scholar]
- 167.Turnbaugh P et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006). [DOI] [PubMed] [Google Scholar]
- 168.Schwiertz A et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 18, 190–195 (2009). [DOI] [PubMed] [Google Scholar]
- 169.John GK & Mullin GE The gut microbiome and obesity. Curr. Oncol. Rep. 18, 45 (2016). [DOI] [PubMed] [Google Scholar]
- 170.Ou J et al. Sa1465 Obesity and colon cancer risk: is it the fat? Gastroenterology 142, S–313 (2012). [Google Scholar]
- 171.Asano TK & McLeod RS Dietary fibre for the prevention of colorectal adenomas and carcinomas. Cochrane Database Syst. Rev. 2, CD003430 (2002). [DOI] [PubMed] [Google Scholar]
- 172.Schatzkin A et al. Lack of effect of a low-fat, high-fiber diet on the recurrence of colorectal adenomas. N. Engl. J. Med. 342, 1149–1155 (2000). [DOI] [PubMed] [Google Scholar]
- 173.Burkitt DP Diseases of the alimentary tract and western diets. Pathol. Microbiol. (Basel) 39, 177–186 (1971). [DOI] [PubMed] [Google Scholar]
- 174.Lanza E et al. High dry bean intake and reduced risk of advanced colorectal adenoma recurrence among participants in the polyp prevention trial. J. Nutr. 136, 1896–1903 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Slavin JL Position of the American Dietetic Association: health implications of dietary fiber. J. Am. Diet Assoc. 108, 1716–1731 (2008). [DOI] [PubMed] [Google Scholar]
- 176.U.S. Department of Health and Human Services and U.S. Department of Agriculture. 2015–2020 Dietary Guidelines for Americans. 8th Edition. http://health.gov/dietaryguidelines/2015/guidelines/ (2015)
- 177.Scientific Advisory Committee on Nutrition. SACN Carbohydrates and Health Report. https://www.gov.uk/government/publications/sacn-carbohydrates-and-health-report (2015)
- 178.Australian National Health and Medical Research Council and the New Zealand Ministry of Health. Nutrient Reference Values for Australia and New Zealand Including Recommended Dietary Intakes. https://www.nrv.gov.au/nutrients/dietary-fibre (2006)
- 179.EFSA Panel on Dietetic Products, Nutrition, and Allergies. Scientific opinion on dietary reference values for carbohydrates and dietary fibre. EFSA Journal 8, 1462 (2010). [Google Scholar]
- 180.O’Keefe SJ The colon as a metabolic organ. S. Afr. Med. J. 84, 376–377 (1994). [PubMed] [Google Scholar]
- 181.O’Keefe SJARP Walker Lecture. Food and the gut. S.Afr. Med. J. 85, 261–268 (1995). [PubMed] [Google Scholar]
- 182.O’Keefe SJ The African way of life and colon cancer risk. Am. J. Gastroenterol. 96, 3220–3221 (2001). [DOI] [PubMed] [Google Scholar]
- 183.O’Keefe SJ Nutrition and colonic health: the critical role of the microbiota. Curr. Opin. Gastroenterol. 24, 51–58 (2008). [DOI] [PubMed] [Google Scholar]
- 184.O’Keefe SJ, Kidd M, Espitalier-Noel G & Owira P Rarity of colon cancer in Africans is associated with low animal product consumption, not fiber. Am. J. Gastroenterol. 94, 1373–1380 (1999). [DOI] [PubMed] [Google Scholar]
- 185.O’Keefe SJ et al. Products of the colonic microbiota mediate the effects of diet on colon cancer risk. J. Nutr. 139, 2044–2048 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Park H et al. A high-fat diet increases angiogenesis, solid tumor growth, and lung metastasis of CT26 colon cancer cells in obesity-resistant BALB/c mice. Mol. Carcinog. 51, 869–880 (2012). [DOI] [PubMed] [Google Scholar]
- 187.Coussens LM & Werb Z Inflammation and cancer. Nature 420, 860–867 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]