

Abstract
Although mucus is a normal product of the tracheobronchial tree, some diseases of the respiratory tract are characterized by unusually thick (inspissated) forms of mucus that accumulate within the airways. These are known as mucus plugs. The pathologic composition of these plugs is surprisingly diverse and, in many cases, correlates with distinctive clinical, radiologic, and bronchoscopic findings. The best-known conditions that involve mucus plugs are allergic bronchopulmonary aspergillosis, plastic bronchitis, and asthma. Other lung diseases occasionally associated with plugs within the airways include Aspergillus tracheobronchitis, hyper-IgE syndrome, exogenous lipoid pneumonia, pulmonary alveolar proteinosis, and chronic eosinophilic pneumonia. In this review, we describe and illustrate the bronchoscopic, pathologic, and imaging findings in respiratory disorders characterized by mucus plugs or plugs composed of other similar materials. Recognition of the characteristic appearance and differential diagnosis of mucus plugs will hopefully facilitate diagnosis and management of these diseases.
Key Words: airway and airspace diseases, bronchoscopic appearance, bronchoscopy, mucoid pseudotumor, mucus plugging
Abbreviations: ABPA, allergic bronchopulmonary aspergillosis; AT, Aspergillus tracheobronchitis; CEP, chronic eosinophilic pneumonia; GM-CSF, granulocyte-macrophage colony-stimulating factor; HIES, hyper-IgE syndrome; PAP, pulmonary alveolar proteinosis; PB, plastic bronchitis
Mucus is a complex mixture of mucin glycoproteins (mucins), water, ions, proteins, and lipids. Its precise composition varies depending on environmental and pathophysiologic conditions. The structure and composition of mucus and mucin glycoproteins have been extensively reviewed in the literature.1 The appearance and odor of sputum has traditionally been used in clinical practice to help identify specific underlying lung conditions. Examples of the appearance of sputum as a clinical clue include currant jelly sputum in Klebsiella infection, brown flakes in aspergillosis, and green sputum in infections caused by Pseudomonas organisms. Examples of odor as a diagnostic clue include the rotten egg odor associated with anaerobic infections and the pungent smell often present in Pseudomonas infections.
The composition of mucus may be altered by pathologic processes that cause stagnation of flow and blockage within the tracheobronchial tree. Mucus that is abnormally thick in consistency (ie, inspissated) and plugs the airway is known as a mucus plug. Consistency and flow of mucus can also be affected not only by pathologic structural abnormalities but also other factors, such as medications with anticholinergic properties. Mucus plugs can partially or completely obstruct one or more airways and cause serious consequences, including atelectasis and recurrent infection. Impacted mucus can also produce a bronchial cast, which is a semisolid occlusion within a bronchus that takes the shape of the airway within which it formed. This type of mucus plug can produce a mass-like appearance on chest imaging that has been termed “mucoid pseudotumor.”2 Mucus plugs that are apparent on imaging and on bronchoscopy can constitute diagnostic clues in certain airway diseases. An important concept in the generation of mucus plugs is the interaction of airway epithelium to external antigens, which can initiate secretagogue-mediated activation and upregulation of secretion of mucin. Patients with chronic airway diseases, such as asthma, cystic fibrosis, and COPD, have increased baseline levels of mucin, and acute exacerbations of disease further contribute to this effect. Secretion of excess mucin catalyzes the formation of biofilm, which in turn contributes to the establishment of pathogens such as Pseudomonas aeruginosa and Klebsiella pneumoniae in structurally affected airways.1
In this review, we focus on diseases of the respiratory tract in which mucus plugs within the airways are a major manifestation of the disease process. We also include disorders in which mucus is not the primary airway-filling material, but bronchoscopically visible mucus plugs can be occasionally encountered. The aim of this review is to familiarize pulmonologists with the differential diagnosis of mucus plugs within the airways and outline the approach to the diagnosis based on imaging, bronchoscopic, and pathologic findings.
Diseases Characterized by Classic Mucin-Containing Mucus Plugs
Allergic Bronchopulmonary Aspergillosis
Aspergillus is a ubiquitous saprophytic fungus that causes a number of distinct syndromes involving the respiratory tract, one of which is allergic bronchopulmonary aspergillosis (ABPA), a well-recognized clinicopathologic entity caused by an allergic-type hypersensitivity response to the presence of fungal hyphae within the airways. This hypersensitivity response is rich in eosinophils and is characterized pathologically by a combination of mucoid impaction of bronchi, bronchiolocentric eosinophil-rich granulomas, and eosinophils within peribronchiolar alveoli.3, 4 Bronchocentric granulomatosis is an inflammatory process that was once thought to be a distinct entity. However, it has become apparent over the years that most examples of this process either occur in patients with ABPA or represent bronchiolocentric granulomatous inflammation in mycobacterial or fungal infections. In contrast, mucoid impaction of bronchi can occur as an independent finding in patients who do not meet other criteria for ABPA. Similarly, pathologic features of eosinophilic pneumonia can occur as part of ABPA, but also in a variety of settings unrelated to ABPA. The complex inter-relationship among these three eosinophil-mediated inflammatory processes has been extensively reviewed.4, 5
Although Aspergillus fumigatus is responsible for most cases of ABPA, other fungi are occasionally implicated in allergic bronchopulmonary fungal disease, including Pseudallescheria boydii, Curvularia species, Torulopsis glabrata, Cryptococcus neoformans, Fusarium vasinfectum, Penicillium species, and other Aspergillus species.6
Most cases of ABPA occur in patients with asthma,7, 8 but it has also been reported in patients with cystic fibrosis9 and in individuals without asthma.10 It is usually diagnosed on the basis of clinical findings.11, 12 In most instances, ABPA presents as a chronic respiratory illness; only rarely does it manifest as acute respiratory failure caused by unilateral lung collapse.13 When mucoid impaction occurs in ABPA, it can obstruct the main bronchi and cause atelectasis or consolidation, compromising respiratory status. In individuals with asthma, the combination of mucoid impaction and airway inflammation is often symptomatic, causing hemoptysis, constitutional symptoms (eg, weight loss, fatigue), and mass-like infiltrates on chest imaging. This presentation can easily be confused for malignancy or pulmonary tuberculosis.7 Clues to the correct diagnosis include a history of asthma, expectoration of mucus plugs, and eosinophilia in the peripheral blood.
On imaging, ABPA is characterized by central bronchiectasis and perihilar mucus plugging. CT scans typically reveal normal or high-density mucus impaction (ie, impaction that appears denser than paraspinal skeletal muscle) (Fig 1). The reason for the high attenuation is not entirely clear, but it has been hypothesized that it is related to calcium and metal salts present in the mucus.14, 15 The distribution of mucus plugs in ABPA differs from their distribution in plastic bronchitis (PB). In ABPA, mucus plugs tend to involve the upper lobes and segmental bronchi, whereas in PB they generally affect the lower lobes and central airways.15 Obstruction of lobar bronchi causes the classic finger-in-glove sign on chest radiography (Fig 2) and can cause lobar collapse (Fig 3). Bronchial casts can be visualized by flexible bronchoscopy, but rigid bronchoscopy and/or CryoProbe (H & O Equipments, Inc) may be required to remove tenacious secretions that do not respond to suction or forceps. In patients with cystic fibrosis, recombinant human deoxyribonuclease may facilitate removal of mucus plugs.9
Figure 1.

A, B, High attenuation mucus (arrows) seen on CT scan of the chest in (A) mediastinal and (B) lung windows.
(Adapted with permission from Agarwal et al.14)
Figure 2.
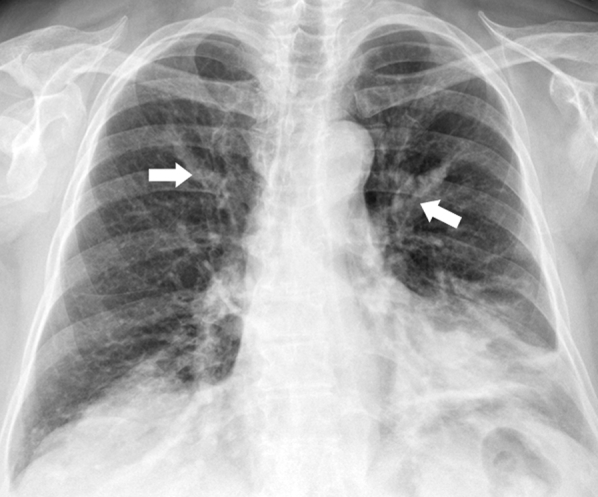
Chest radiograph shows finger-in-glove sign (arrows) from left upper lobe mucus impaction in a patient with allergic bronchopulmonary aspergillosis.
Figure 3.

A, B, Chest radiograph shows (A) collapsed right upper lobe from mucus impaction in a patient with allergic bronchopulmonary aspergillosis (pretreatment) and (B) resolution of right upper lobe collapse (postbronchoscopic treatment).
The distinctive pathologic features of mucus plugs in ABPA and mucoid impaction of bronchi have been well described.3, 5, 16 The microscopic appearance of the mucin is known as allergic mucin. In contrast with normal mucus or mucus in other pathologic conditions such as uncomplicated asthma, allergic mucin has a lamellated microscopic appearance at low magnification (Fig 4), caused by alternating layers of mucin and fibrin that contain numerous viable, degenerating, and necrotic eosinophils (Fig 5) and Charcot-Leyden crystals (Fig 6).4, 17 Charcot-Leyden crystals are rhomboid or bipyramidal structures composed of lysophospholipase, an enzyme synthesized by eosinophils.18 Their presence is a marker of eosinophil-rich inflammation, but is otherwise nonspecific. Small numbers of degenerating fungal hyphae may be demonstrable within the mucus plugs of ABPA by histopathology or cultures. In contrast with invasive aspergillosis, fungal hyphae do not invade the airway wall or lung parenchyma in ABPA.
Figure 4.
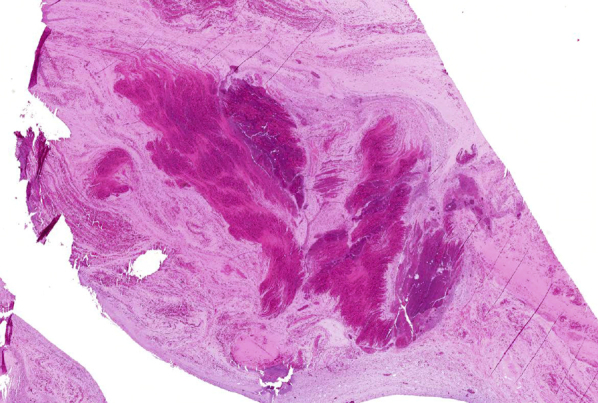
Microscopic appearance of mucus plugs in mucoid impaction of bronchi and allergic bronchopulmonary aspergillosis (allergic mucin), showing characteristic lamellated appearance under low magnification (40×).
Figure 5.
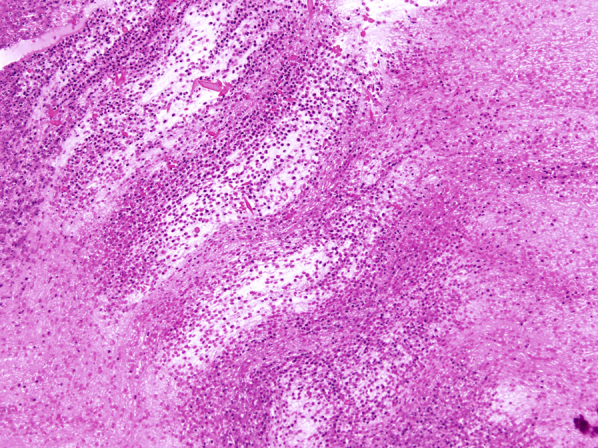
Microscopic appearance of mucus plugs in mucoid impaction of bronchi and allergic bronchopulmonary aspergillosis (allergic mucin) under high magnification, showing alternating layers of mucin (pale) and fibrin (dark). Numerous eosinophils and Charcot-Leyden crystals are present in both layers (magnification, 200×).
Figure 6.
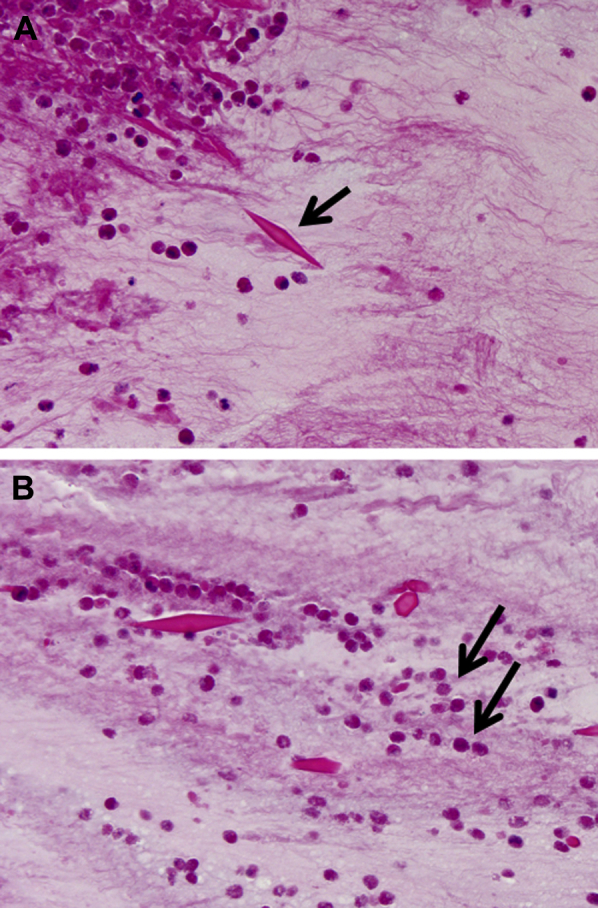
A, B, Charcot-Leyden crystals within allergic mucin in allergic bronchopulmonary aspergillosis. Bipyramidal appearance (arrow). B, Degenerating eosinophils are present in the background (arrows). Charcot-Leyden crystals can be found in any inflammatory process that contains numerous eosinophils (magnification, 400×).
In addition to evacuation of the obstructing mucus plugs, patients with ABPA require long-term corticosteroid therapy, which is currently thought to be the mainstay of management.6 Antifungal treatment is considered adjunctive. Obstructive symptoms can recur after completion of the initial treatment course. Therefore, these patients must be closely followed and may require repeat bronchoscopy and reinitiation of systemic therapy.
Plastic Bronchitis
Many terms have been used to describe PB, including pseudomembranous bronchitis, mucoid impaction, cast bronchitis, fibrinous bronchitis, bronchitis fibroplastica, and Hoffman bronchitis.19, 20 PB is a rare disorder that usually occurs in children (average age, 6 years; range, 1-17 years), but it has been described in adults as well. The exact incidence of PB remains unclear because it is a rare condition, so the diagnosis is likely missed in some patients.21 It is characterized by branching mucoid bronchial casts that obstruct the tracheobronchial tree, causing respiratory distress.20 The casts have been classified into two groups based on the presumed underlying pathophysiology. Type I casts (inflammatory) consist predominantly of eosinophil-rich cellular infiltrates and occur primarily in children with asthma or cystic fibrosis. Type II casts (acellular) are hypocellular and are encountered in patients with a history of surgically corrected cyanotic congenital heart disease.22 Acellular casts have also been associated with chyle leakage from the endobronchial lymphatic system.23 Broadly speaking, the pathology and pathogenesis of type I casts overlap with asthma and ABPA, whereas type II casts appear to represent a more distinct entity.
Clinically, patients with PB present with wheezing, low-grade fever, upper respiratory tract infection, dyspnea, expectoration of tree-like casts, and frequent hemoptysis.22 The condition can mimic foreign body aspiration or status asthmaticus, and urgent bronchoscopy may be required for diagnosis and treatment.24 Imaging studies typically reveal atelectasis, lung infiltrates, hyperinflation of the contralateral lung, or bronchial cutoff. In advanced cases, the imaging characteristics may resemble those of bronchiectasis.25 For patients in respiratory distress in whom PB is suspected, emergent bronchoscopy is recommended. The mucus plugs of PB are described as bronchial casts; that is, they take the shape of the bronchi that they fill. Although the tree-like gross appearance can be striking, the histologic appearance is underwhelming. The casts in the more distinctive form of PB (type II) usually contain mucin, fibrin, and a few lymphocytes and macrophages (Fig 7). Figure 8 shows a bronchoscopic view of complete obstruction of the right main bronchus by a bronchial cast in a patient with PB.
Figure 7.
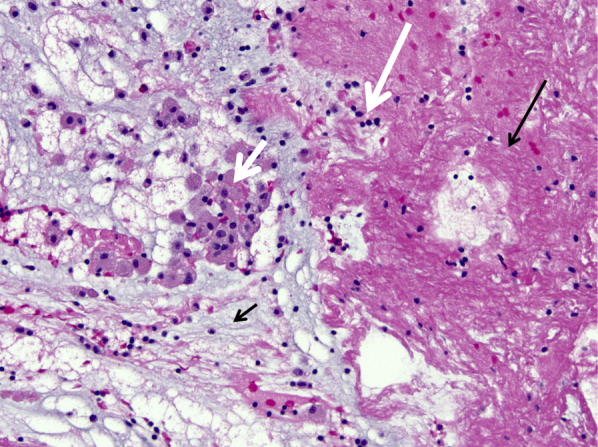
Low magnification of mucus plug in plastic bronchitis containing mucin (short black arrow), fibrin (long black arrow), macrophages (short white arrow), and a few lymphocytes (long white arrow) (magnification, 200×).
Figure 8.
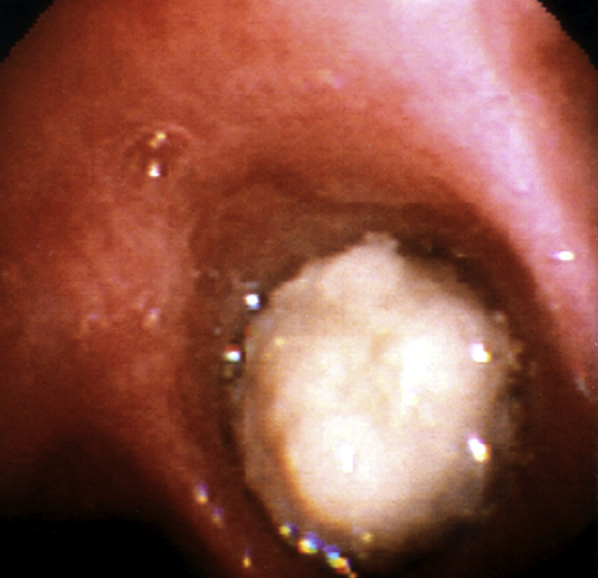
Bronchoscopic view of complete obstruction of the right main bronchus with a large cast in a patient with plastic bronchitis.
(Image courtesy of Dr Daniel Sterman.)
Removal of these large, rubbery mucus plugs presents a unique therapeutic challenge because they are usually too soft to grasp with forceps and too thick to suction. After removal, the plug is immersed in saline or formalin to help demonstrate its characteristic branching pattern, which forms a perfect cast of the bronchus from which it was removed. The presence of blood in the gross specimen should not alter the differential diagnosis (Fig 9).24 Previous publications have described the use of other therapies for mucus clearance, including mucolytics, chest physiotherapy, intratracheal recombinant human deoxyribonuclease, hydration, and aerosolized acetylcysteine.26 The use of anti-inflammatory therapy with corticosteroids and macrolides has also been reported, with varying degrees of success.27 In cases of PB in which lymphatic abnormalities are causative, a low-fat, medium-chain triglyceride diet or thoracic duct ligation may be helpful.28 These patients must be closely followed to allow for early bronchoscopic intervention and prevention of further morbidity.
Figure 9.

A, Left-sided airway cast removed via rigid bronchoscopy from a patient with plastic bronchitis. B, Airway cast resembling the entire bronchial tree, removed via rigid bronchoscopy from a patient with plastic bronchitis.
(Adapted with permission from Sriratanaviriyakul et al.79)
Asthma
Mucus plugging of the bronchi is one of the most common autopsy findings in patients with fatal status asthmaticus (Fig 10). A major contributing cause of fatal asthma is obstruction of the airway lumen by an exudate composed of mucus and cell debris. In one autopsy series, patients who died of asthma showed a greater degree of luminal occlusion by mucus and cells than patients who died suddenly without pulmonary disease.29 Mucus plugs can occur not only in pediatric patients with severe asthma, but also in children in remission from asthma,30 and they can obstruct small or large airways. Similar to children, adults suffering from asthma can also be affected. Although multilobe involvement is common, obstructions are more likely to occur in the right lung.30 Mucus plugs have also been reported in patients with asthma who have undergone bronchial thermoplasty (Fig 11).31 The etiology of mucus plugging in asthma is probably multifactorial. Marked hypertrophy and hyperplasia of goblet cells in the airways, upregulation of the MUC5AC gene, and stimulation of secretions are known contributors to production of excessive mucus.32, 33
Figure 10.

A, B, Autopsy specimen from a patient with fatal asthma. A, Bronchial wall is characterized by thick mucoid secretions within the lumen, marked eosinophilic infiltration, smooth muscle hypertrophy, and thickening of the bronchial basement membrane. B, Gross specimen shows the mucus plugging in the airway (arrow).
(Adapted with permission from Mohiuddin et al.81)
Figure 11.

A-C, Mucus plug obstructing the right lower and middle lobe bronchi (A) after bronchial thermoplasty in a patient with asthma. B, Histologic examination showed fibrin mixed with some mucus, scattered inflammatory cells and (C) gross examination showed a bronchus-shaped mucus plug.
(Adapted with permission from Facciolongo et al.31)
Mucus plugs in patients with asthma are composed of mucin, fibrin, and variable numbers of inflammatory cells, including macrophages, neutrophils, eosinophils, and T-lymphocytes (Figs 4, 5, 10, 11).34 Microscopically, they lack the lamellated appearance and large numbers of eosinophils that characterize the mucus plugs of ABPA and mucoid impaction of bronchi.16 Curschmann spirals are microscopic structures that may be found in cytologic samples prepared from the sputum of individuals with asthma. Shaped like spirals, they are composed of mucus that has originated from distal small airways. Although Curschmann spirals are traditionally associated with asthma, they have also been found in asymptomatic smokers, in patients with lung cancer or chronic bronchitis, and in individuals exposed to urban atmospheric pollution.35 The mucus of asthmatics may also contain Charcot-Leyden crystals (see section on ABPA for complete discussion and Fig 6) and three-dimensional clusters of benign ciliated bronchial cells known as Creola bodies. Mucus plugs are only detectable on radiography in roughly 2.2% of patients with asthma.30 However, coaxial chest CT scan may reveal mucus impaction along with other radiographic abnormalities, findings that correlate with increasing severity of asthma, and decreasing FEV1.36 As noted previously, the presence of high-density mucus in patients with asthma should prompt evaluation for ABPA.14
Although mucus plugs are involved in airway obstruction in status asthmaticus, mucolytic therapies have not been effective in improving outcomes. Current guidelines set forth by the National Asthma Education and Prevention Program37 do not recommend chest physical therapy or mucolytics, such as N-acetyl cysteine, in patients with status asthmaticus. Nevertheless, glucocorticoids and anticholinergic agents may help regulate hypersecretion of mucus.34
Diseases That May Feature Mucus Plugs or Airway Plugs of Other Types
Aspergillus Tracheobronchitis
Aspergillus tracheobronchitis (AT) is a relatively rare but severe form of aspergillosis involving the tracheobronchial tree (Fig 12). The burden of disease is increased in patients who are immunocompromised or who have neutropenia and in those undergoing lung transplantation or cytotoxic chemotherapy. AT is classified into three histopathologic patterns: obstructive, ulcerative, and pseudomembranous.38, 49 The obstructive and pseudomembranous forms—which are associated with mucus plugging in the tracheobronchial tree40—are subsequently discussed in greater detail.
Figure 12.
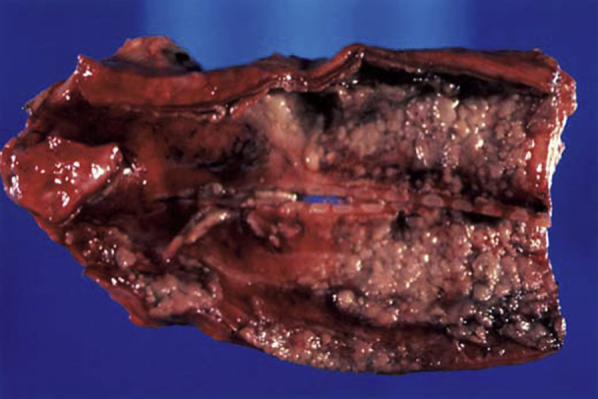
Gross autopsy specimen of the trachea reveals inflammatory exudates in a patient with aspergillus tracheobronchitis.
(Adapted with permission from Minai and Mehta.40)
The obstructive form of AT was first recognized in patients with AIDS who expectorated thick mucus plugs containing Aspergillus but had little or no inflammation and no evidence of invasive or allergic disease.39 Obstructive AT has also been reported to cause middle lobe atelectasis in heart transplant recipients.41 Symptoms are often insidious, and bronchoscopy is required for diagnosis and removal of the obstructing plugs. Mucoid obstruction may progress to invasive tracheobronchitis in immunocompromised hosts,39, 41 and lung transplant patients are particularly susceptible to airway complications resulting from Aspergillus infection. Cerceo et al42 reported the presence of large, fibrinous, Aspergillus-laden mucus plugs obstructing the central airway associated with a rapid reduction in FEV1. Administration of steroids and itraconazole resolved these complications.42 Involvement of the major lobar bronchi with complete lobar collapse was recently reported in a liver transplant recipient (Figs 13, 14).43
Figure 13.

A-D, CT scan of the chest in a liver transplant recipient with Aspergillus tracheobronchitis shows complete collapse of the left lower lobe in the (A, B) mediastinal and (C, D) lung windows.
(Adapted with permission from Panchabhai et al.43)
Figure 14.

A, Bronchoscopic view of completely obstructed left main bronchus from large mucus plug. B, C, The plug was subsequently removed. D, Distal mucus plugs were still visible in the lobar bronchi after initial removal and were later aspirated.
(Adapted with permission from Panchabhai et al.43)
The pathologic features of the mucus plugs associated with obstructive AT are not well described. They are thought to be full of Aspergillus hyphae and eosinophils, but invasion is limited to superficial layers, differentiating this condition from classic invasive aspergillosis. Although colonization of the airways and anastomotic sites occurs in 50% to 70% of lung transplant recipients,42 eosinophils, large mucus plugs, and invasion of the main bronchi with associated lobar collapse are uncommon.
Pseudomembranous AT is a form of invasive aspergillosis characterized by extensive pseudomembrane formation and sloughing of necrotic epithelium in the airways. It is more common in patients with hematologic malignancies than in lung transplant recipients.44 Patients with hematologic malignancies who have pseudomembranous AT frequently present with fever, inspiratory wheezing, dyspnea, and nonproductive cough.45 Severe pseudomembranous AT can cause atelectasis, airway obstruction, and acute respiratory failure.46 Bronchoscopy facilitates collection of microbiologic and pathologic specimens, which is essential for the diagnosis.45 Bronchoscopic aspirates frequently contain abundant fungal hyphae and mucosal slough, and tissue invasion is apparent on biopsy specimens. Removal and biopsy of the pseudomembrane carries the risk of fatal bleeding because of the propensity of Aspergillus for vascular invasion. In patients with thrombocytopenia, invasion may be exaggerated.47
In patients with inconclusive endobronchial specimens, additional tissue specimens can be obtained via endobronchial ultrasound-guided transbronchial needle aspiration of lung lesions adjacent to the airways.48 The Infectious Diseases Society of America49 recommends voriconazole for primary treatment of invasive AT. Alternatives to voriconazole include liposomal amphotericin B, caspofungin, micafungin, posaconazole, or itraconazole. Bronchoscopic removal of pseudomembranes and endobronchial masses may be a useful adjunct to systemic therapy because antifungals may be limited in their ability to penetrate the fungal masses. However, extreme caution should be exercised because bronchoscopic removal carries a high risk of severe bleeding.50
Hyper-IgE Syndromes
Hyper-IgE syndromes (HIES) are a group of primary immunodeficiency syndromes associated with significantly elevated levels of IgE (usually > 2,000 International Unit/mL), recurrent skin infections, eczema, and pulmonary infections. Two distinct clinical forms have been described: autosomal-dominant HIES and autosomal-recessive HIES.51 Several mutations have been associated with HIES; mutations of signal transducer and activator of transcription 3 and DOCK8 are the most common.
The clinical manifestations of HIES vary depending on the underlying mutation, but recurrent pulmonary and cutaneous infections are the most common clinical manifestations.52 Staphylococcus aureus and Streptococcus pneumoniae are the most commonly isolated bacteria associated with HIES. Recurrent pneumonia may lead to long-term sequelae, such as pneumatoceles and bronchiectasis.53, 54 Pneumatoceles are not seen in autosomal-recessive HIES; however, recurrent pneumonia is common.51 Patients with pneumatoceles or bronchiectasis are also prone to secondary infection with Aspergillus and gram-negative bacteria, and opportunistic infections such as Pneumocystis pneumonia have also been reported. Mucus plugging of the airways is often seen in these recurrent secondary infections. Bronchoscopy is indicated for microbiologic diagnosis and for airway clearance.
Pulmonary complications are the most common cause of death in patients with HIES. In a postmortem study of six patients with autosomal-dominant HIES, all had pneumonia at the time of death, with Aspergillus and Pseudomonas being the predominant causes of infection (66% and 33%, respectively).55 Mucus plugs obstructing the airway with resultant pneumonia is a common clinical presentation of HIES (Fig 15).
Figure 15.

A-D, CT scan of the chest shows mucus plugging resulting in obstruction of multiple airways and distal consolidation in a patient with hyper-IgE syndrome.
Treatment of HIES is primarily supportive. Antibiotic prophylaxis against S. aureus with sulfamethoxazole and secondary antifungal prophylaxis is recommended and has been shown to decrease the incidence of recurrent infection.53, 56 Immunomodulatory therapy with intravenous immunoglobulin has been tried with variable success. Surgical intervention is often required; in a recent analysis, 22% of patients required lobectomy.53 Surgical intervention should be pursued with caution, however, because it carries a high risk of complications, such as bronchopleural fistulae and impaired lung expansion.
Lipoid Pneumonia
Two clinically and pathologically distinct processes are characterized by accumulation of lipid or lipid-like material within the lungs. Exogenous lipoid pneumonia is a rare but well-characterized disorder caused by aspiration of exogenous oily material (ie, mineral, vegetable, or animal oil) into the lungs.57 The most commonly implicated materials are liquid paraffin and mineral oil, both of which are used for treatment of constipation.58 However, a wide variety of other oily substances have been reported to cause the disease, including petroleum-based lubricants (eg, Vaseline), oily nose drops, laxatives, spray lubricants, and lip gloss.59 Risk factors that predispose to aspiration include gastroesophageal reflux disease and neurologic or psychiatric illness. In one case, exogenous lipoid pneumonia was reported in a commercial abalone diver who presumably aspirated aerosolized mineral oil contained in the unfiltered air generated from his surface air compressor.57, 60, 61
The word exogenous refers to the fact that the lipid-like (lipoid) material in this condition is derived from materials extraneous to the human body. In contrast, so-called endogenous lipoid pneumonia is a common but nonspecific consequence of airway obstruction or narrowing by a wide variety of causes, including neoplasms, infection, inflammation, and other etiologies.62 The lipid in endogenous lipoid pneumonia is derived from cell membranes of macrophages, whose egress from the lung is impeded by narrowed airways. The accumulated macrophages eventually disintegrate, and the lipid from their cell membranes is ingested by other macrophages, which then take on what has been described as a foamy appearance. Accumulation of large numbers of foamy macrophages within the alveoli imparts the characteristic golden hue to the lung that has prompted terms such as golden pneumonia and cholesterol pneumonia. A similar golden material may fill the airways.
A large proportion of patients with exogenous lipoid pneumonia are asymptomatic at presentation. In symptomatic individuals, the clinical presentation is usually insidious; persistent cough, dyspnea, fever, and weight loss are the most common symptoms. Airspace disease is the most prevalent findings on chest CT scan. Other patterns include ground glass nodules, solid nodules, and crazy paving. Lung involvement is usually bilateral and multilobar, with a predilection for the lower lobes.60, 63 A density measurement between −30 and −150 Hounsfield units suggests the presence of fat in the lesion and may point toward this diagnosis.60
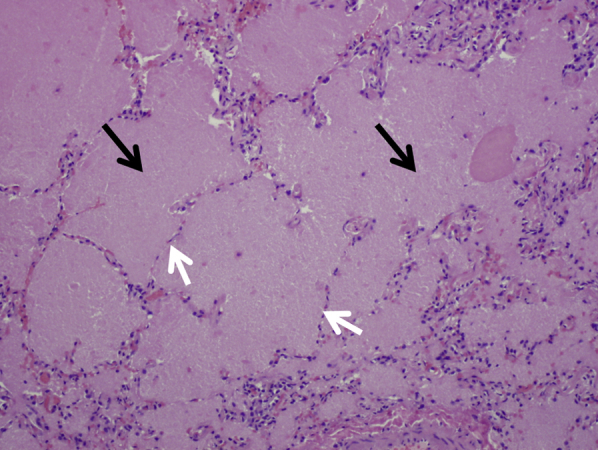
As reported by Gondouin et al,60 BAL and transbronchial biopsy help confirm the diagnosis in most patients with exogenous lipoid pneumonia. BAL fluid in exogenous lipoid pneumonia is often milky or oily (Fig 16)60, 64 and contains foamy macrophages with large (coarse) vacuoles. The pathologic appearance in transbronchial or surgical lung biopsies is confirmatory. It is characterized by the presence of foamy macrophages within the airspaces (alveolar lumens) and/or interstitium, containing variably sized coarse vacuoles within their cytoplasm (Fig 17). In contrast, the foamy macrophages seen in endogenous lipoid pneumonia contain fine vacuoles of uniform size that are present exclusively within airspaces (Fig 18). Although the vacuoles of exogenous lipoid pneumonia are remnants of lipid-like substances, the lipid itself is removed by processing in formalin-fixed paraffin-embedded sections.60, 64 Varying degrees of interstitial fibrosis are often present in exogenous lipoid pneumonia, and a giant cell reaction to the lipoid material in the interstitium is common. Cytologic profiles are nonspecific; lymphocytic, neutrophilic, and mixed profiles have been reported. In contrast with formalin-fixed paraffin-embedded transbronchial lung biopsies, BAL fluid samples can be subjected to fat stains, such as oil red O, which should be requested if the diagnosis is suspected clinically. Bronchoscopy may show plugs within the airways (Fig 19), occasionally associated with aspirated food particles.
Figure 16.
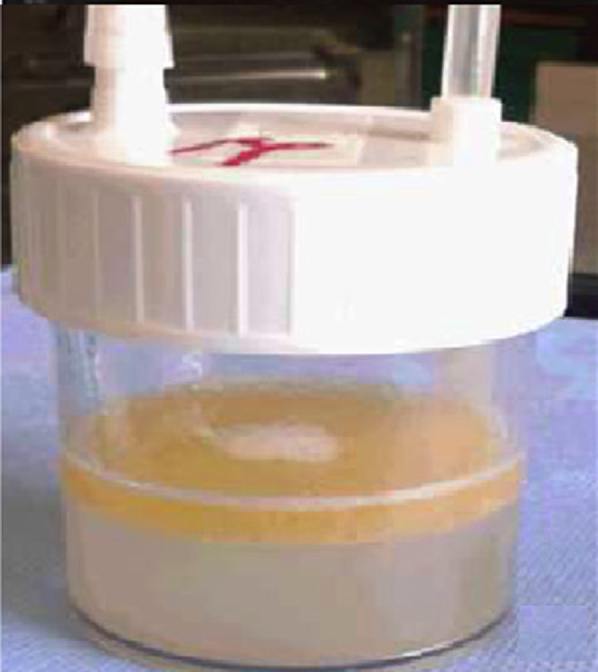
Gross appearance of BAL fluid in exogenous lipoid pneumonia in a patient taking Lorenzo’s oil.
(Adapted with permission from Majori et al.82)
Figure 17.
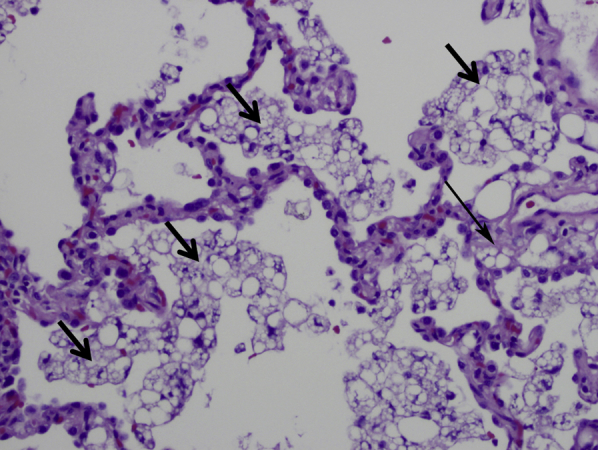
Microscopic appearance of exogenous lipoid pneumonia. Macrophages containing coarse vacuoles of varying sizes fill the airspaces (short arrows). Similar macrophages are focally present within the interstitium (long arrow). This appearance is pathognomonic (magnification, 200×).
Figure 18.
Microscopic appearance of endogenous lipoid pneumonia. Macrophages containing fine uniform vacuoles fill the airspaces (arrows). This is a common and nonspecific pathologic finding in lung specimens (magnification, 200×).
Figure 19.

A, Bronchoscopic view of exogenous lipoid pneumonia with airway plugs. B, Photograph of airway plugs removed from a patient with exogenous lipoid pneumonia.
Identifying and discontinuing use of the causative agent is the cornerstone of therapy.58 There are anecdotal reports of successful treatment of exogenous lipoid pneumonia with corticosteroids and whole lung lavage.65
Pulmonary Alveolar Proteinosis
Pulmonary alveolar proteinosis (PAP) is caused by the abnormal accumulation of surfactant phospholipoprotein within alveoli. This accumulation is thought to be caused by failure of alveolar macrophages to clear surfactant from the lungs. PAP presents as one of three distinct forms: congenital, secondary, or acquired.66 The congenital form is characterized by an acute onset of rapidly progressive respiratory distress immediately after birth.67 Secondary PAP is secondary to conditions that reduce the number of alveolar macrophages or inhibit their function, such as some hematologic malignancies and infections.66 Acquired PAP (commonly known as autoimmune PAP) is by far the most common form of PAP in adulthood. It usually affects middle-aged individuals; 90% of cases have an autoimmune etiology and are associated with antibodies directed against granulocyte-macrophage colony-stimulating factor (GM-CSF). PAP is more prevalent in men and has a strong association with cigarette smoking.68, 69
The onset of acquired PAP is usually insidious with slowly progressive dyspnea and cough; however, acute respiratory failure has also been reported.70 There is often a prolonged latency before diagnosis, and approximately 30% of patients are asymptomatic at presentation.69
High-resolution CT scan is useful in the initial evaluation of patients with suspected PAP. Radiographic abnormalities are often out of proportion to symptoms (ie, they are surprisingly dramatic despite the associated mild symptomatology). The classic radiologic finding is a crazy-paving pattern on high-resolution CT scan, which manifests as bilateral ground-glass opacities superimposed on a background of smooth interlobular and intralobular septal thickening. However, the crazy-paving pattern can be seen in several other pulmonary diseases and is therefore not specific for PAP.71 Although PAP is defined by filling of the alveoli, the tracheobronchial tree may also be affected (Fig 20).72 Dense lipoproteinaceous material plugging the distal small airways has resulted in segmental atelectasis in some cases.
Figure 20.

A-D, Bronchoscopic images show mucoid plugs within the airways in a patient with pulmonary alveolar proteinosis. Note the white-yellow, thick appearance.
(Image courtesy of Dr Abdul Hamid Alraiyes.)
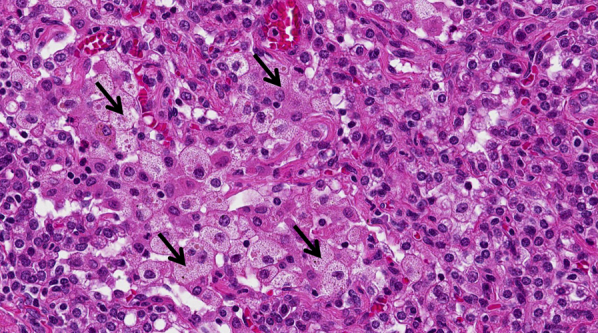
Diagnosis of PAP is greatly facilitated by BAL and transbronchial lung biopsy. On gross examination, BAL fluid in PAP is milky and opaque. After it has settled, it separates into two layers: thick sediment at the bottom and a clear supernatant at top. On light microscopy, the airspaces (the lumens of the alveoli) are filled with a pathognomonic eosinophilic amorphous material (Fig 21). This material often contains acicular spaces and dense eosinophilic globules. It is usually, but not invariably, periodic acid-Schiff stain-positive and diastase-resistant.71 Alveolar septa are normal in most cases, despite what one would expect from the interstitial appearance of the disease on imaging. Anti-GM-CSF antibody titers can be measured in BAL fluid and correlate with severity of disease.73
Figure 21.
Microscopic findings in a patient with pulmonary alveolar proteinosis. Granular eosinophilic material fills the alveoli (black arrows). Alveolar septa (white arrows) are normal (magnification, 40×).
Therapeutic lung lavage is an established treatment modality for PAP, resulting in clinical, symptomatic, and radiographic improvement and reduced mortality. Lavage needs to be repeated in most patients (median of twice per patient).71 Exogenous GM-CSF has recently been reported as a therapy for autoimmune PAP; however, it is associated with less clinical improvement than with therapeutic lung lavage and can take up to 8 to 12 weeks to appear.71
Chronic Eosinophilic Pneumonia
Chronic eosinophilic pneumonia (CEP) is an idiopathic inflammatory disorder whose pathologic hallmark is the accumulation of large numbers of eosinophils within the alveoli. It typically affects patients in their 30s and 40s, many of whom have a history of atopy or asthma. Common symptoms include productive cough, fever, dyspnea, weight loss, and night sweats. Although uncommon, CEP can also present with bronchoconstriction and mucus plugs in the lower lung and has been reported in children and in middle-aged adults.74, 75 The classic radiologic pattern has been described as the “photographic negative of pulmonary edema.”76 However, this pattern is present only in 25% of cases; therefore, its absence does not exclude CEP.76
Mucus plugs occur in some patients with CEP. In these patients, they appear as thick, yellow-white impactions inside the airways. The most likely cause of mucus plugging in these patients is the presence of eosinophil-rich inflammatory infiltrates, conceptually similar to those seen in asthma and ABPA. The diagnosis of CEP can be confirmed by lung biopsy and BAL, both of which can demonstrate large numbers of eosinophils. CEP is exquisitely sensitive to corticosteroid therapy.
Other Conditions
Mucus dysfunction has also been described in patients with panbronchiolitis and in those with immunodeficiencies (eg, hypogammaglobinemia, HIV, malignancy, transplant recipients). Mucus dysfunction can lead to retention of secretions, mucus plugging, atelectasis, and bronchiectasis. Genetic markers such as MUC5AC and MUC5B abnormalities have been associated with many of these conditions.77 In recent years, newer therapeutic procedures such as transtracheal oxygen catheter placement and bronchoscopic thermoplasty have been shown to produce mucus plugs (Figs 11, 22).31, 78 Diagnosis in such conditions can easily be established with thorough history taking. Bronchoscopically, airway plugs may also be present in patients with infectious pneumonias caused by a variety of organisms; however, these plugs differ histopathologically from mucin-containing plugs (Fig 23)83 and chronic aspiration.
Figure 22.

A, Tracheal cast in situ from a patient who died from complications of mucus plug after insertion of a transtracheal oxygen catheter. Arrows indicate the upper and lower edges of the cast, which filled approximately 90% of the lumen. B, Microscopic image of the tracheal cast showing a distinct marbled pattern of homogenous proteinaceous zones alternating with dark zones of necrotic epithelial cellular nuclei and inflammatory cells.
(Adapted with permission Burton et al.78)
Figure 23.

A-C, Bronchoscopic images show necrotic debris and plugs in the (A) left main, (B) left upper, and (C) left lower lobe bronchi in a patient with community-acquired methicillin-resistant Staphylococcus aureus pneumonia.
(Adapted with permission Panchabhai et al.83)
Bronchoscopic Management of Mucus Plugs
Initial management of mucus plugs involves proper hydration, humidification, bronchodilation, and use of mucolytic agents via nebulization. If these measures fail, flexible bronchoscopy may be required. If performed, flexible bronchoscopy may also provide diagnostic information. Although most plugs are amenable to suction techniques, tenacious mucus plugs can be hard to aspirate, requiring advanced bronchoscopic measures. Cryoadhesion has been used to remove thick mucus plugs adherent to the airways.43, 79 This technique allows an efficient approach to remove tenacious mucus, blood clots, and organic foreign bodies from the endobronchial tree in a minimally invasive fashion. It is of paramount importance that mucus specimens retrieved via bronchoscopy be sent for culture and in-depth pathologic analysis when the diagnosis is unclear.80
Conclusions
This review describes the common respiratory disorders characterized by plugs within the air passages, with an emphasis on mucus plugs. Not all mucus plugs within the large airways are alike; specific clinical, imaging, and pathologic features can help to narrow the differential diagnosis.
Acknowledgments
Financial/nonfinancial disclosures: None declared.
Footnotes
FUNDING/SUPPORT: The authors have reported to CHEST that no funding was received for this study.
References
- 1.Rose M.C., Voynow J.A. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol Rev. 2006;86(1):245–278. doi: 10.1152/physrev.00010.2005. [DOI] [PubMed] [Google Scholar]
- 2.Karasick D., Karasick S., Lally J.F. Mucoid pseudotumors of the tracheobronchial tree in two cases. AJR Am J Roentgenol. 1979;132(3):459–460. doi: 10.2214/ajr.132.3.459. [DOI] [PubMed] [Google Scholar]
- 3.Bosken C.H., Myers J.L., Greenberger P.A., Katzenstein A.L. Pathologic features of allergic bronchopulmonary aspergillosis. Am J Surg Pathol. 1988;12(3):216–222. doi: 10.1097/00000478-198803000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Mukhopadhyay S., Gal A.A. Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med. 2010;134(5):667–690. doi: 10.5858/134.5.667. [DOI] [PubMed] [Google Scholar]
- 5.Katzenstein A.L., Liebow A.A., Friedman P.J. Bronchocentric granulomatosis, mucoid impaction, and hypersensitivity reactions to fungi. Am Rev Respir Dis. 1975;111(4):497–537. doi: 10.1164/arrd.1975.111.4.497. [DOI] [PubMed] [Google Scholar]
- 6.Greenberger P.A., Bush R.K., Demain J.G., Luong A., Slavin R.G., Knutsen A.P. Allergic bronchopulmonary aspergillosis. J Allergy Clin Immunol Pract. 2014;2(6):703–708. doi: 10.1016/j.jaip.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cote C.G., Cicchelli R., Hassoun P.M. Hemoptysis and a lung mass in a 51-year-old patient with asthma. Chest. 1998;114(5):1465–1468. doi: 10.1378/chest.114.5.1465. [DOI] [PubMed] [Google Scholar]
- 8.Scheer B.G., Hutcheson P.S., Lagos J., Wood J., Slavin R.G. Mucoid impaction: a localized form of allergic bronchopulmonary aspergillosis. Allergy Asthma Proc. 2004;25(4):229–232. [PubMed] [Google Scholar]
- 9.Cakir E., Uyan Z.S., Ersu R.H., Karadag B., Karakoc F., Dagli E. Mucoid impaction: an unusual form of allergic bronchopulmonary aspergillosis in a patient with cystic fibrosis. Pediatr Pulmonol. 2006;41(11):1103–1107. doi: 10.1002/ppul.20499. [DOI] [PubMed] [Google Scholar]
- 10.Berkin K.E., Vernon D.R., Kerr J.W. Lung collapse caused by allergic bronchopulmonary aspergillosis in non-asthmatic patients. Br Med J (Clin Res Ed) 1982;285(6341):552–553. doi: 10.1136/bmj.285.6341.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenberger P.A., Patterson R. Diagnosis and management of allergic bronchopulmonary aspergillosis. Ann Allergy. 1986;56(6):444–448. [PubMed] [Google Scholar]
- 12.Rosenberg M., Patterson R., Mintzer R., Cooper B.J., Roberts M., Harris K.E. Clinical and immunologic criteria for the diagnosis of allergic bronchopulmonary aspergillosis. Ann Intern Med. 1977;86(4):405–414. doi: 10.7326/0003-4819-86-4-405. [DOI] [PubMed] [Google Scholar]
- 13.Agarwal R., Aggarwal A.N., Gupta N., Gupta D. A rare cause of acute respiratory failure–allergic bronchopulmonary aspergillosis. Mycoses. 2011;54(4):e223–e227. doi: 10.1111/j.1439-0507.2009.01830.x. [DOI] [PubMed] [Google Scholar]
- 14.Agarwal R., Gupta D., Aggarwal A.N., Saxena A.K., Chakrabarti A., Jindal S.K. Clinical significance of hyperattenuating mucoid impaction in allergic bronchopulmonary aspergillosis: an analysis of 155 patients. Chest. 2007;132(4):1183–1190. doi: 10.1378/chest.07-0808. [DOI] [PubMed] [Google Scholar]
- 15.Borbely B.R., Davies A.L., Jones M. Left lung atelectasis in a smoker. Chest. 1994;105(6):1833–1835. doi: 10.1378/chest.105.6.1833. [DOI] [PubMed] [Google Scholar]
- 16.Jelihovsky T. The structure of bronchial plugs in mucoid impaction, bronchocentric granulomatosis and asthma. Histopathology. 1983;7(2):153–167. doi: 10.1111/j.1365-2559.1983.tb02232.x. [DOI] [PubMed] [Google Scholar]
- 17.Chetty A. Pathology of allergic bronchopulmonary aspergillosis. Front Biosci. 2003;8:e110–e114. doi: 10.2741/945. [DOI] [PubMed] [Google Scholar]
- 18.Ackerman S.J., Liu L., Kwatia M.A. Charcot-Leyden crystal protein (galectin-10) is not a dual function galectin with lysophospholipase activity but binds a lysophospholipase inhibitor in a novel structural fashion. J Biol Chem. 2002;277(17):14859–14868. doi: 10.1074/jbc.M200221200. [DOI] [PubMed] [Google Scholar]
- 19.Jett J.R., Tazelaar H.D., Keim L.W., Ingrassia T.S., 3rd Plastic bronchitis: an old disease revisited. Mayo Clin Proc. 1991;66(3):305–311. doi: 10.1016/s0025-6196(12)61013-1. [DOI] [PubMed] [Google Scholar]
- 20.Werkhaven J., Holinger L.D. Bronchial casts in children. Ann Otol Rhinol Laryngol. 1987;96(1 Pt 1):86–92. doi: 10.1177/000348948709600121. [DOI] [PubMed] [Google Scholar]
- 21.Eberlein M.H., Drummond M.B., Haponik E.F. Plastic bronchitis: a management challenge. Am J Med Sci. 2008;335(2):163–169. doi: 10.1097/MAJ.0b013e318068b60e. [DOI] [PubMed] [Google Scholar]
- 22.Seear M., Hui H., Magee F., Bohn D., Cutz E. Bronchial casts in children: a proposed classification based on nine cases and a review of the literature. Am J Respir Crit Care Med. 1997;155(1):364–370. doi: 10.1164/ajrccm.155.1.9001337. [DOI] [PubMed] [Google Scholar]
- 23.Languepin J., Scheinmann P., Mahut B. Bronchial casts in children with cardiopathies: the role of pulmonary lymphatic abnormalities. Pediatr Pulmonol. 1999;28(5):329–336. doi: 10.1002/(sici)1099-0496(199911)28:5<329::aid-ppul4>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 24.Noizet O., Leclerc F., Leteurtre S. Plastic bronchitis mimicking foreign body aspiration that needs a specific diagnostic procedure. Intensive Care Med. 2003;29(2):329–331. doi: 10.1007/s00134-002-1610-1. [DOI] [PubMed] [Google Scholar]
- 25.Liston S.L., Porto D., Siegel L.G. Plastic bronchitis. Laryngoscope. 1986;96(12):1347–1351. doi: 10.1288/00005537-198612000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Manna S.S., Shaw J., Tibby S.M., Durward A. Treatment of plastic bronchitis in acute chest syndrome of sickle cell disease with intratracheal rhDNase. Arch Dis Child. 2003;88(7):626–627. doi: 10.1136/adc.88.7.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schultz K.D., Oermann C.M. Treatment of cast bronchitis with low-dose oral azithromycin. Pediatr Pulmonol. 2003;35(2):139–143. doi: 10.1002/ppul.10196. [DOI] [PubMed] [Google Scholar]
- 28.Shah S.S., Drinkwater D.C., Christian K.G. Plastic bronchitis: Is thoracic duct ligation a real surgical option? Ann Thorac Surg. 2006;81(6):2281–2283. doi: 10.1016/j.athoracsur.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 29.Kuyper L.M., Pare P.D., Hogg J.C. Characterization of airway plugging in fatal asthma. Am J Med. 2003;115(1):6–11. doi: 10.1016/s0002-9343(03)00241-9. [DOI] [PubMed] [Google Scholar]
- 30.Maxwell G.M. The problem of mucus plugging in children with asthma. J Asthma. 1985;22(3):131–137. doi: 10.3109/02770908509073131. [DOI] [PubMed] [Google Scholar]
- 31.Facciolongo N., Menzella F., Lusuardi M. Recurrent lung atelectasis from fibrin plugs as a very early complication of bronchial thermoplasty: a case report. Multidiscip Respir Med. 2015;10(1):9. doi: 10.1186/s40248-015-0002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aikawa T., Shimura S., Sasaki H., Ebina M., Takishima T. Marked goblet cell hyperplasia with mucus accumulation in the airways of patients who died of severe acute asthma attack. Chest. 1992;101(4):916–921. doi: 10.1378/chest.101.4.916. [DOI] [PubMed] [Google Scholar]
- 33.Evans C.M., Kim K., Tuvim M.J., Dickey B.F. Mucus hypersecretion in asthma: causes and effects. Curr Opin Pulm Med. 2009;15(1):4–11. doi: 10.1097/MCP.0b013e32831da8d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rogers D.F. Pulmonary mucus: Pediatric perspective. Pediatr Pulmonol. 2003;36(3):178–188. doi: 10.1002/ppul.10322. [DOI] [PubMed] [Google Scholar]
- 35.Cenci M., Giovagnoli M.R., Alderisio M., Vecchione A. Curschmann's spirals in sputum of subjects exposed daily to urban environmental pollution. Diagn Cytopathol. 1998;19(5):349–351. doi: 10.1002/(sici)1097-0339(199811)19:5<349::aid-dc7>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 36.Harmanci E., Kebapci M., Metintas M., Ozkan R. High-resolution computed tomography findings are correlated with disease severity in asthma. Respiration. 2002;69(5):420–426. doi: 10.1159/000064018. [DOI] [PubMed] [Google Scholar]
- 37.National Asthma Education and Prevention Program Expert Panel Report 3 (EPR-3): Guidelines for the Diagnosis and Management of Asthma-Summary Report 2007. J Allergy Clin Immunol. 2007;120(5 suppl):S94–S138. doi: 10.1016/j.jaci.2007.09.043. [DOI] [PubMed] [Google Scholar]
- 38.Denning D.W. Commentary: unusual manifestations of aspergillosis. Thorax. 1995;50(7):812–813. doi: 10.1136/thx.50.7.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Denning D.W., Follansbee S.E., Scolaro M., Norris S., Edelstein H., Stevens D.A. Pulmonary aspergillosis in the acquired immunodeficiency syndrome. N Engl J Med. 1991;324(10):654–662. doi: 10.1056/NEJM199103073241003. [DOI] [PubMed] [Google Scholar]
- 40.Minai O.A., Mehta A.C. A lung-transplant recipient with infiltrates. Cleve Clin J Med. 2001;68(2):172–173. doi: 10.3949/ccjm.68.2.172. [DOI] [PubMed] [Google Scholar]
- 41.Hummel M., Schuler S., Hempel S., Rees W., Hetzer R. Obstructive bronchial aspergillosis after heart transplantation. Mycoses. 1993;36(11-12):425–428. doi: 10.1111/j.1439-0507.1993.tb00733.x. [DOI] [PubMed] [Google Scholar]
- 42.Cerceo E., Kotloff R.M., Hadjiliadis D. Central airways obstruction due to Aspergillus fumigatus after lung transplantation. J Heart Lung Transplant. 2009;28(5):515–519. doi: 10.1016/j.healun.2009.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Panchabhai T.S., Bandyopadhyay D., Alraiyes A.H., Mehta A.C., Almeida F.A. A 60-year-old woman with cough, dyspnea, and atelectasis 19 years after liver transplant. Chest. 2015;148(4):e122–e125. doi: 10.1378/chest.15-0388. [DOI] [PubMed] [Google Scholar]
- 44.Tasci S., Glasmacher A., Lentini S. Pseudomembranous and obstructive Aspergillus tracheobronchitis - optimal diagnostic strategy and outcome. Mycoses. 2006;49(1):37–42. doi: 10.1111/j.1439-0507.2005.01180.x. [DOI] [PubMed] [Google Scholar]
- 45.van Assen S., Bootsma G.P., Verweij P.E., Donnelly J.P., Raemakers J.M. Aspergillus tracheobronchitis after allogeneic bone marrow transplantation. Bone Marrow Transplant. 2000;26(10):1131–1132. doi: 10.1038/sj.bmt.1702679. [DOI] [PubMed] [Google Scholar]
- 46.Routsi C., Kaltsas P., Bessis E., Rontogianni D., Kollias S., Roussos C. Airway obstruction and acute respiratory failure due to Aspergillus tracheobronchitis. Crit Care Med. 2004;32(2):580–582. doi: 10.1097/01.CCM.0000110724.86196.3B. [DOI] [PubMed] [Google Scholar]
- 47.Garofano S.A., Stover D.E., Freeberg G.W., Klimstra D.S. Necrotizing aspergillus tracheobronchitis: a case associated with fatal hemorrhage following endobrachial biopsy. Journal of Bronchology. 1994;1(4):299–303. [Google Scholar]
- 48.Casal R.F., Adachi R., Jimenez C.A., Sarkiss M., Morice R.C., Eapen G.A. Diagnosis of invasive aspergillus tracheobronchitis facilitated by endobronchial ultrasound-guided transbronchial needle aspiration: a case report. J Med Case Rep. 2009;3:9290. doi: 10.1186/1752-1947-3-9290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walsh T.J., Anaissie E.J., Denning D.W. Treatment of aspergillosis: clinical practice guidelines of the Infectious Diseases Society of America. Clin Infect Dis. 2008;46(3):327–360. doi: 10.1086/525258. [DOI] [PubMed] [Google Scholar]
- 50.Berlinger N.T., Freeman T.J. Acute airway obstruction due to necrotizing tracheobronchial aspergillosis in immunocompromised patients: a new clinical entity. Ann Otol Rhinol Laryngol. 1989;98(9):718–720. doi: 10.1177/000348948909800911. [DOI] [PubMed] [Google Scholar]
- 51.Freeman A.F., Holland S.M. Clinical manifestations, etiology, and pathogenesis of the hyper-IgE syndromes. Pediatr Res. 2009;65(5 Pt 2):32R–37R. doi: 10.1203/PDR.0b013e31819dc8c5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moin M., Farhoudi A., Movahedi M. The clinical and laboratory survey of Iranian patients with hyper-IgE syndrome. Scand J Infect Dis. 2006;38(10):898–903. doi: 10.1080/00365540600740470. [DOI] [PubMed] [Google Scholar]
- 53.Chandesris M.O., Melki I., Natividad A. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine (Baltimore) 2012;91(4):e1–e19. doi: 10.1097/MD.0b013e31825f95b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grimbacher B., Holland S.M., Gallin J.I. Hyper-IgE syndrome with recurrent infections–an autosomal dominant multisystem disorder. N Engl J Med. 1999;340(9):692–702. doi: 10.1056/NEJM199903043400904. [DOI] [PubMed] [Google Scholar]
- 55.Freeman A.F., Kleiner D.E., Nadiminti H. Causes of death in hyper-IgE syndrome. J Allergy Clin Immunol. 2007;119(5):1234–1240. doi: 10.1016/j.jaci.2006.12.666. [DOI] [PubMed] [Google Scholar]
- 56.Kuruvilla M., de la Morena M.T. Antibiotic prophylaxis in primary immune deficiency disorders. J Allergy Clin Immunol Pract. 2013;1(6):573–582. doi: 10.1016/j.jaip.2013.09.013. [DOI] [PubMed] [Google Scholar]
- 57.Kupeli E., Khemasuwan D., Tunsupon P., Mehta A.C. “Pills” and the air passages: a continuum. Chest. 2015;147(1):242–250. doi: 10.1378/chest.14-0531. [DOI] [PubMed] [Google Scholar]
- 58.Marchiori E., Zanetti G., Mano C.M., Hochhegger B. Exogenous lipoid pneumonia. Clinical and radiological manifestations. Respir Med. 2011;105(5):659–666. doi: 10.1016/j.rmed.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 59.Simmons A., Rouf E., Whittle J. Not your typical pneumonia: a case of exogenous lipoid pneumonia. J Gen Intern Med. 2007;22(11):1613–1616. doi: 10.1007/s11606-007-0280-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gondouin A., Manzoni P., Ranfaing E. Exogenous lipid pneumonia: a retrospective multicentre study of 44 cases in France. Eur Respir J. 1996;9(7):1463–1469. doi: 10.1183/09031936.96.09071463. [DOI] [PubMed] [Google Scholar]
- 61.Kizer K.W., Golden J.A. Lipoid pneumonitis in a commercial abalone diver. Undersea Biomed Res. 1987;14(6):545–552. [PubMed] [Google Scholar]
- 62.Genereux G.P. Lipids in the lungs: radiologic-pathologic correlation. J Can Assoc Radiol. 1970;21(1):2–15. [PubMed] [Google Scholar]
- 63.Marchiori E., Zanetti G., Mano C.M., Irion K.L., Daltro P.A., Hochhegger B. Lipoid pneumonia in 53 patients after aspiration of mineral oil: comparison of high-resolution computed tomography findings in adults and children. J Comput Assist Tomogr. 2010;34(1):9–12. doi: 10.1097/RCT.0b013e3181a9ec9f. [DOI] [PubMed] [Google Scholar]
- 64.Lauque D., Dongay G., Levade T., Caratero C., Carles P. Bronchoalveolar lavage in liquid paraffin pneumonitis. Chest. 1990;98(5):1149–1155. doi: 10.1378/chest.98.5.1149. [DOI] [PubMed] [Google Scholar]
- 65.Chang H.Y., Chen C.W., Chen C.Y. Successful treatment of diffuse lipoid pneumonitis with whole lung lavage. Thorax. 1993;48(9):947–948. doi: 10.1136/thx.48.9.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Trapnell B.C., Whitsett J.A., Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 25 2003;349(26):2527–2539. doi: 10.1056/NEJMra023226. [DOI] [PubMed] [Google Scholar]
- 67.Khan A., Agarwal R. Pulmonary alveolar proteinosis. Respir Care. 2011;56(7):1016–1028. doi: 10.4187/respcare.01125. [DOI] [PubMed] [Google Scholar]
- 68.Goldstein L.S., Kavuru M.S., Curtis-McCarthy P., Christie H.A., Farver C., Stoller J.K. Pulmonary alveolar proteinosis: clinical features and outcomes. Chest. 1998;114(5):1357–1362. doi: 10.1378/chest.114.5.1357. [DOI] [PubMed] [Google Scholar]
- 69.Inoue Y., Trapnell B.C., Tazawa R. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med. 2008;177(7):752–762. doi: 10.1164/rccm.200708-1271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gacouin A., Le Tulzo Y., Suprin E. Acute respiratory failure caused by secondary alveolar proteinosis in a patient with acute myeloid leukemia: a case report. Intensive Care Med. 1998;24(3):265–267. doi: 10.1007/s001340050563. [DOI] [PubMed] [Google Scholar]
- 71.Seymour J.F., Presneill J.J. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med. 2002;166(2):215–235. doi: 10.1164/rccm.2109105. [DOI] [PubMed] [Google Scholar]
- 72.Prakash U.B., Barham S.S., Carpenter H.A., Dines D.E., Marsh H.M. Pulmonary alveolar phospholipoproteinosis: experience with 34 cases and a review. Mayo Clin Proc. 1987;62(6):499–518. doi: 10.1016/s0025-6196(12)65477-9. [DOI] [PubMed] [Google Scholar]
- 73.Venkateshiah S.B., Yan T.D., Bonfield T.L. An open-label trial of granulocyte macrophage colony stimulating factor therapy for moderate symptomatic pulmonary alveolar proteinosis. Chest. 2006;130(1):227–237. doi: 10.1378/chest.130.1.227. [DOI] [PubMed] [Google Scholar]
- 74.Cakir E., Aksoy F., Cakir F.B., Ertem T. Chronic eosinophilic pneumonia with mucous plugs in a child. Pediatr Pulmonol. 2010;45(10):1040–1042. doi: 10.1002/ppul.21299. [DOI] [PubMed] [Google Scholar]
- 75.Xie L.X., Mo G.X., Chen L.A., Liu Y.N. Chronic eosinophilic pneumonia with mucous plugs: case report. Chin Med J (Engl) 2006;119(3):262–264. [PubMed] [Google Scholar]
- 76.Jederlinic P.J., Sicilian L., Gaensler E.A. Chronic eosinophilic pneumonia. A report of 19 cases and a review of the literature. Medicine (Baltimore) 1988;67(3):154–162. doi: 10.1097/00005792-198805000-00002. [DOI] [PubMed] [Google Scholar]
- 77.Fahy J.V., Dickey B.F. Airway mucus function and dysfunction. N Engl J Med. 2010;363(23):2233–2247. doi: 10.1056/NEJMra0910061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Burton G.G., Wagshul F.A., Henderson D., Kime S.W. Fatal airway obstruction caused by a mucous ball from a transtracheal oxygen catheter. Chest. 1991;99(6):1520–1523. doi: 10.1378/chest.99.6.1520. [DOI] [PubMed] [Google Scholar]
- 79.Sriratanaviriyakul N., Lam F., Morrissey B.M., Stollenwerk N., Schivo M., Yoneda K.Y. Safety and clinical utility of flexible bronchoscopic cryoextraction in patients with non-neoplasm tracheobronchial obstruction: a retrospective chart review. J Bronchology Interv Pulmonol. 2015;22(4):288–293. doi: 10.1097/LBR.0000000000000203. [DOI] [PubMed] [Google Scholar]
- 80.Soyer T., Yalcin S., Emiralioglu N. Use of serial rigid bronchoscopy in the treatment of plastic bronchitis in children. J Pediatr Surg. 2016;51(10):1640–1643. doi: 10.1016/j.jpedsurg.2016.03.017. [DOI] [PubMed] [Google Scholar]
- 81.Mohiuddin M., Zacharisen M.C., Poulos C., Levy M.B. Asthma deaths outside the hospital in an urban community from 2004 to 2008. Ann Allergy Asthma Immunol. 2012;108(6):412–417. doi: 10.1016/j.anai.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 82.Majori M., Scarascia A., Anghinolfi M., Pisi R., Gnetti L., Casalini A.G. Lipoid pneumonia as a complication of Lorenzo's oil therapy in a patient with adrenoleukodystrophy. J Bronchology Interv Pulmonol. 2014;21(3):271–273. doi: 10.1097/LBR.0000000000000084. [DOI] [PubMed] [Google Scholar]
- 83.Panchabhai T.S., Khabbaza J.E., Raja S., Mehta A.C., Hatipoglu U. Extracorporeal membrane oxygenation and toilet bronchoscopy as a bridge to pneumonectomy in severe community-acquired methicillin-resistant Staphylococcus aureus pneumonia. Ann Thorac Med. 2015;10(4):292–294. doi: 10.4103/1817-1737.164298. [DOI] [PMC free article] [PubMed] [Google Scholar]