

SUMMARY
Hallmarks of aging that negatively impact health include weight gain and reduced physical fitness, which can increase insulin resistance and risk for many diseases including type 2 diabetes. The underlying mechanism(s) for these phenomena is poorly understood. Here we report that aging increases DNA breaks and activates DNA-dependent protein kinase (DNA-PK) in skeletal muscle, which suppresses mitochondrial function, energy metabolism and physical fitness. DNA-PK phosphorylates threonines 5 and 7 of HSP90α, decreasing its chaperone function for clients such as AMP-activated protein kinase (AMPK), which is critical for mitochondrial biogenesis and energy metabolism. Decreasing DNA-PK activity increases AMPK activity and prevents weight gain, decline of mitochondrial function and physical fitness in middle aged mice and protects against type 2 diabetes. Therefore, DNA-PK is one of the drivers of the metabolic and fitness decline during aging, which make staying lean and physically fit difficult and increase susceptibility to metabolic diseases.
Keywords: DNA-PK, HSP90α, AMPK, aging, calorie restriction, type 2 diabetes, mitochondria, exercise, skeletal muscle, obesity, insulin sensitivity
Graphical abstract
INTRODUCTION
One of the aging-associated metabolic changes is the loss of mitochondrial content and function in tissues such as skeletal muscle (Barazzoni et al., 2000; Petersen et al., 2003; Short et al., 2005). Since mitochondria convert nutrients to energy and heat, the mitochondrial decline is relevant to aging-associated decline in metabolic rate and exercise capacity. Lee et. al. (Lee et al., 2010) reported that the rise of mitochondrial reactive oxygen species (ROS) with aging causes a decline in mitochondrial function in skeletal muscle. This can lead to metabolic dysfunctions such as insulin resistance, which can lead to chronic diseases such as type 2 diabetes.
Accumulating evidence indicates that the aging-associated decline in skeletal muscle activity of AMP-activated protein kinase (AMPK), a key regulator of mitochondrial function and energy balance (Hardie, 2007), plays an important role in the metabolic decline associated with aging (Koonen et al., 2010; Lee et al., 2010; Qiang et al., 2007; Reznick et al., 2007). AMPK has numerous functions including stimulation of glucose uptake, fat oxidation, energy production and mitochondrial biogenesis (Ruderman et al., 2013). Increased AMPK activity decreases visceral fat (Narkar et al., 2008) and increases mitochondrial biogenesis and energy production in skeletal muscle resulting in improved physical fitness (Zong et al., 2002). On the other hand, AMPK-deficiency in skeletal muscle leads to mitochondrial loss, impaired glucose uptake and exercise intolerance (O’Neill et al., 2011). Metformin, the most commonly used type 2 diabetes drug acts in part by AMPK activation (Zhou et al., 2001). However, the molecular mechanism by which aging decreases AMPK activity in skeletal muscle is poorly understood.
DNA-PK (Jackson, 1997) is activated by DNA double-stranded breaks (DSBs). It is a trimeric complex composed of the catalytic subunit, DNA-PKcs, and the Ku70/80 heterodimer. DNA-PK mediates non-homologous end-joining (NHEJ), which joins programmed DSBs created during V(D)J recombination and class switching recombination in lymphocytes (Critchlow and Jackson, 1998). As a result, severe combined immune deficiency (SCID) mice (Blunt et al., 1996), which carry a leaky mutation in DNA-PKcs, have impaired lymphocyte development. As a DNA DSB sensor, DNA-PK has some unusual properties. For example, DNA-PK is very abundant: it is estimated that HeLa cells contain approximately 100,000 copies of DNA-PKcs per cell (Anderson and Carter, 1996), far in excess of what is probably needed for NHEJ. In addition, DNA-PKcs is present not only in the nucleus but also in the cytoplasm (Huston et al., 2008). In agreement with this, evidence is mounting that DNA-PK has functions beyond genetic stability. One such function is in metabolism: DNA-PK phosphorylates transcription factor USF-1 and promotes fatty acid synthesis in response to insulin (Wong et al., 2009).
One of the proteins DNA-PK interacts with and phosphorylates is HSP90α (Lees-Miller and Anderson, 1989), one of the two isoforms of HSP90 (α and β). HSP90 is unique among chaperone proteins in that it binds to and folds only a small fraction of the total proteome called “clients,” the majority of which are protein kinases (Li and Buchner, 2013). DNA-PK phosphorylates Thr5,7 (T5,7) in the extreme N-terminus (ENT) region of HSP90α (aa. 1–11, Figure S1) (Lees-Miller and Anderson, 1989; Quanz et al., 2012; Solier et al., 2012), but DNA-PK does not phosphorylate HSP90β (Lees-Miller and Anderson, 1989). The effect of T5,7 phosphorylation on HSP90α function is not known.
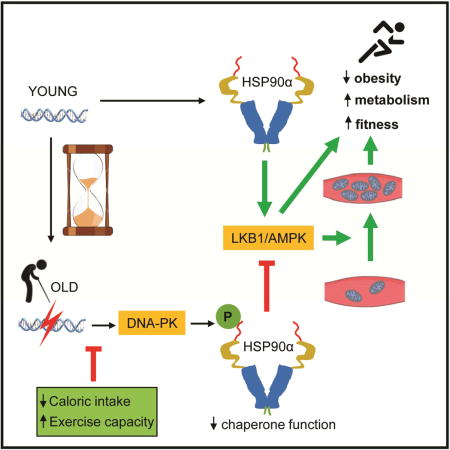
Here, we show that T5,7 phosphorylation of HSP90α, which increases with aging in skeletal muscle, disrupts the HSP90α-client complex. AMPK (Taipale et al., 2012) and its upstream activator kinase LKB1 are known HSP90 clients. Preventing T5,7 phosphorylation, either by mutating T5,7 or by genetic loss of DNA-PKcs increases AMPK activity in skeletal muscle. Loss of DNA-PK activity protects against diet-induced obesity and insulin resistance and against loss of mitochondria and physical fitness at older age in mice. Therefore, DNA-PK promotes metabolic and physical decline in older age.
RESULTS
Aging increases DNA DSBs and DNA-PK activity
A naturally occurring condition that may increase DNA DSBs and potentially activate DNA-PK is aging. In order to determine if aging is associated with increased DNA DSBs in skeletal muscle, we isolated skeletal muscle from young (3 mo) and old (25 mo) mice and visualized γ-H2AX (Rogakou et al., 1998), a phosphorylated form of histone H2AX that is a marker for DNA DSBs (Figure 1A). We found that skeletal muscle from old mice has more γ-H2AX, suggesting that aging increases DNA DSBs in skeletal muscle.
Figure 1. Aging increases DNA DSBs and DNA-PK activity.
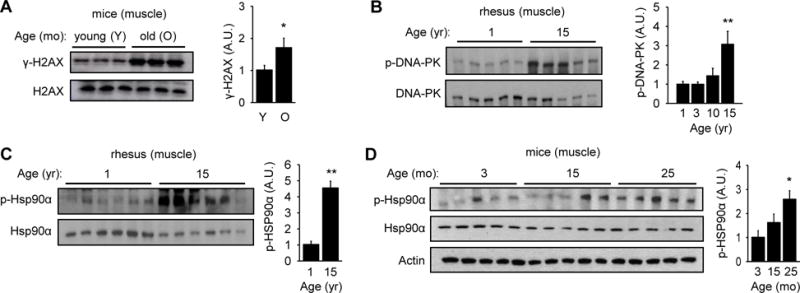
(A) H2AX phosphorylation (γ-H2AX) in skeletal muscle of young (3 mo) and old (25 mo) mice (in arbitrary units) (n=10 each group). Quantification is shown on the right. All values are expressed as mean ± s.e.m. Mann-Whitney Test: *, p<0.05 between the two age groups. (B) DNA-PK autophosphorylation (S2056) in skeletal muscle of rhesus (in arbitrary units) (n=5 each age group). Quantification is shown on the right. Mann-Whitney Test: **, p<0.01 compared to the youngest group. (C) p-HSP90α in skeletal muscle of 1 and 15 yr old rhesus (in arbitrary units) (n=6 per group). Quantification is shown on the right. Mann-Whitney Test: **, p<0.01 compared to the youngest group. (D) p-HSP90α in mice skeletal muscle according to age (in arbitrary units) (n=5 each group). Quantification is shown on the right. Mann-Whitney Test: *, p<0.05 compared to the youngest group.
We then examined the association of aging and DNA-PK activity in two different organisms: rhesus macaques and mice. We isolated skeletal muscle (gastrocnemius) from healthy rhesus macaques ranging in age from 1 to 15 yr, which is equivalent to 3 to 45 yr in human age. The tissue was isolated from fasted animals to exclude food intake as a variable. We detected DNA-PK activity by visualizing autophosphorylation of S2056 (p-DNA-PK) in DNA-PKcs (Chen et al., 2005). In all cases, the level phosphorylation was normalized to the total protein level. The levels of p-DNA-PK were similar at 1, 3 and 10 yr (image not shown), but rose significantly at 15 yr (Figure 1B). The antibody specific for p-DNA-PK, which was raised against human DNA-PKcs, was too weakly cross reacting for detection of mouse p-DNA-PK (data not shown).
Consistent with the dramatic increase in p-DNA-PK at middle aged, T5,7-HSP90α (p-HSP90α) was significantly higher in middle aged macacque skeletal muscle (Figure 1C). Similarly, p-HSP90α was significantly higher in skeletal muscle from old mice compared to that in young mice (Figure 1D). These findings indicate that both DNA-PK activity and p-HSP90α are elevated in skeletal muscle of older rhesus monkeys and mice. Interestingly, p-HSP90α did not increase with aging in the lung, indicating that aging does not increase p-HSP90α in all tissues (Figure S2).
DNA-PK-mediated phosphorylation of HSP90α triggers release of clients
Although HSP90 is more abundant in the cytoplasm, there is some HSP90 in the nucleus (van Bergen en Henegouwen et al., 1987). In order to determine the cellular compartment to which phosphorylated p-HSP90α is localized, we purified the cytoplasmic and nuclear fractions before and after ionizing radiation (IR), which generates DNA DSBs and activates DNA-PK (Figure 2A). We found a detectable basal level of p-HSP90α in the cytoplasm and nucleus, but after IR, p-HSP90α increased dramatically in both compartments. Moreover, IR increased nuclear accumulation of HSP90α but not HSP90β.
Figure 2. DNA-PK-mediated phosphorylation of HSP90α triggers release of clients.
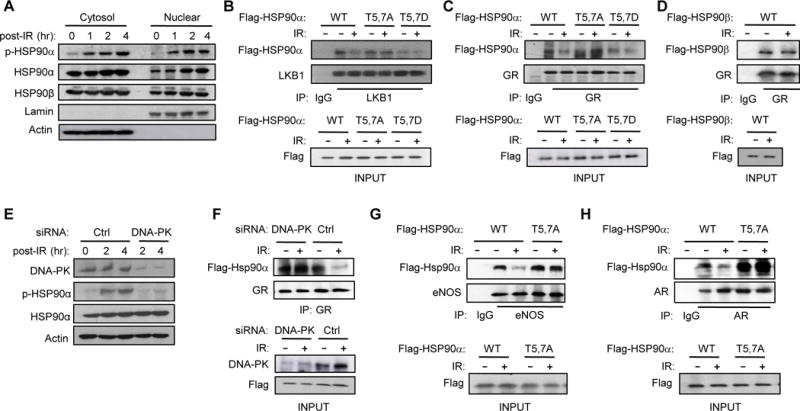
(A) T5,7 are phosphorylated in both cytoplasmic and nuclear HSP90α in response to DNA DSBs induced by IR (15 Gy) in C2C12 myotubes. (B) C2C12 myotubes were transfected with vectors encoding Flag-tagged WT or mutant HSP90α. Interaction between mutant HSP90α and endogenous LKB1 was visualized by immunoprecipitating with anti-LKB1 antibody one hr after IR and immunoblotting with anti-Flag antibody. (C) Interaction between Flag-tagged WT or mutant HSP90α and endogenous GR one hr after IR was visualized by immunoprecipitating with anti-GR antibody and immunoblotting with anti-Flag antibody. (D) Interaction between Flag-tagged HSP90β and GR one hr after IR was visualized by immunoprecipitating with anti-GR antibody and immunoblotting with anti-Flag antibody. (E) C2C12 myotubes transfected with siRNA for either Ctrl or DNA-PKcs were treated with IR (4Gy) and the expression level of p-HSP90α and DNA-PK were visualized by immunoblotting. (F) C2C12 myotubes transfected with a vector encoding WT HSP90α and siRNA for either Ctrl or DNA-PKcs were treated with IR and harvested one hour later. Interaction between GR and HSP90α is shown. (G and H) The effect of IR on the interaction between HSP90α mutants and either endogenous eNOS (G) or AR (H) in C2C12 myotubes is shown.
Based on a previous report showing that the ENT region may regulate HSP90 chaperone function by affecting the HSP90-client complex formation (Pullen and Bolon, 2011), we examined whether T5,7 phosphorylation affects HSP90α-client interaction. To test this, we transiently-transfected C2C12 myotubes with vectors encoding Flag-tagged WT, T5,7A (T→A) mutant and phosphomimetic T5,7D (T→D) mutant HSP90α, treated them with IR and examined the interaction of the HSP90α mutants with the clients liver kinase B1 (LKB1) and glucocorticoid receptor (GR). Co-immunoprecipitation studies showed that IR released LKB1 from WT HSP90α, but not from T5,7A HSP90α (Figure 2B). T5,7D HSP90α bound poorly to LKB1 even in the absence of IR. Similarly, IR released GR from WT HSP90α, but not from T5,7A HSP90α (Figure 2C); T5,7D HSP90α also bound poorly to GR even in the absence of IR. Unlike the HSP90α-GR interaction, the HSP90β-GR interaction was not affected by IR (Figure 2D). To demonstrate that the IR-induced release of GR from HSP90α was mediated by DNA-PK-dependent phosphorylation, we performed co-immunoprecipitation between GR and HSP90α after knocking-down DNA-PKcs with siRNA. As shown in Figure 2E, DNA-PKcs siRNA significantly reduced p-HSP90α and prevented IR from disrupting the GR-HSP90α interaction (Figure 2F). The T5,7A mutations also blunted IR-induced release of other HSP90 clients such as endothelial nitric oxide synthase (eNOS) (Figure 2G) and the androgen receptor (AR) (Figure 2H) from HSP90α. Taken together, these findings indicate that DNA-PK-mediated phosphorylation of HSP90α can disrupt HSP90α-client complexes.
Inhibition of DNA-PK activates AMP-activated protein kinase
Since AMPK (Taipale et al., 2012) and its upstream activator kinase LKB1 (Nony et al., 2003) are HSP90 clients, we investigated whether DNA-PK and phosphorylation of HSP90α mediate the aging-related decline in AMPK activity. Although AMPKα2 (the dominant AMPK in skeletal muscle) is an HSP90 client, its interaction with HSP90 is weak to begin with (background intensity) (Taipale et al., 2012), making it difficult to see further decrease in interaction after IR. However, to confirm that AMPK is a client of HSP90α, we knocked-down HSP90α with siRNA and visualized the level of AMPK in C2C12 myotubes. Decreased chaperone function of HSP90 often results in decreased levels of client proteins due to aggregation and/or destabilization. Consistent with the notion that AMPK is a client of HSP90α, HSP90α siRNA decreased AMPK levels (Figure 3A top and bottom left panels) without affecting its mRNA levels (Figure 3A bottom right panel), and treatment with the HSP90 inhibitor 17-AAG also decreased AMPK levels (Figure 3B). We also investigated whether the T5,7 phosphorylation affects the AMPK-HSP90α interaction by transiently expressing WT and T5,7A HSP90α. As shown in Figure 3C, AMPK interaction with T5,7A HSP90α was stronger than with WT HSP90α and consistent with this, p-AMPK (T172), a marker of AMPK activity, was higher with T5,7A HSP90α compared with WT HSP90α (Figure 3D). These findings suggest that phosphorylation of T5,7 decreases HSP90α-AMPK complex formation and AMPK activity.
Figure 3. Inhibition of DNA-PK activates AMP-activated protein kinase.
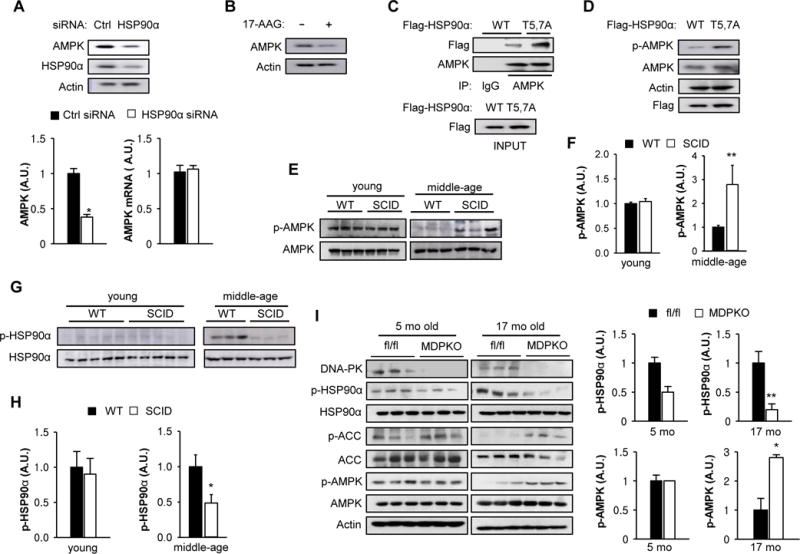
(A) C2C12 myotubes were transfected with a siRNA for either control (Ctrl) or HSP90α and the expression level of HSP90α and AMPK were visualized by immunoblotting. Quantification is shown on the bottom left. All values are expressed as mean ± s.e.m. Mann-Whitney Test: *, p<0.05. (Bottom Right) C2C12 myotubes were transfected with a siRNA for either Ctrl or HSP90α and AMPKα mRNA was measured by using real-time PCR. All values are given as mean ± s.e.m. (B) C2C12 myotubes were treated with HSP90 inhibitor (1 μM 17-AAG) and the expression level of AMPK was visualized by immunoblotting. (C) C2C12 myotubes were transfected with vectors encoding Flag-tagged WT or T5,7A HSP90α. Interaction between mutant HSP90α and endogenous AMPK was visualized by immunoprecipitating with Flag antibody. (D) C2C12 myotubes were transfected with vectors encoding either WT or T5,7A HSP90α and the expression levels of Flag-tagged HSP90α and p-AMPK (T172) were visualized by immunoblotting. (E) AMPK phosphorylation (p-AMPK) in resting muscle isolated from young (3–4 months old) and middle aged (12–14 mo) WT and SCID mice. Three representative samples for each group are shown. (F) Quantification of p-AMPK in young (n=3 per genotype) and middle aged muscle (n=6 per genotype) from WT and SCID mice. Mann-Whitney Test: **, p<0.01. (G) HSP90α phosphorylation in resting skeletal muscle isolated from young and middle aged WT and SCID mice. (H) Quantification of p-HSP90α in young and middle aged muscle (n=7 per genotype). Mann-Whitney Test: *, p<0.05. (I) AMPK activation (p-AMPK, p-ACC1) and HSP90α phosphorylation in resting skeletal muscle isolated from young (5 months old) and old (17 months old) fl/fl and muscle specific DNA-PK knockout (MDPKO) mice. Quantification is shown on the right. Mann-Whitney Test: *, p<0.05: **, p<0.01.
We then investigated whether DNA-PK affects AMPK activity in vivo. For this purpose, we used severe combined immunodeficiency (SCID) mice, which carry a leaky mutation in DNA-PKcs (Blunt et al., 1996) but have a normal lifespan (Bailey et al., 1999). We visualized p-AMPK in skeletal muscle of young (3–4 mo) and middle aged (12–14 mo) WT and SCID mice (Figures 3E and F). In young SCID mice, p-AMPK was similar to that in WT mice but in middle aged SCID mice, p-AMPK was higher than that in WT mice. In line with this observation, p-HSP90α was also decreased in middle aged SCID muscle relative to WT muscle (Figures 3G and H). To confirm that the DNA-PK-AMPK link was cell autonomous in vivo, we generated a conditional knockout of DNA-PKcs in skeletal muscle (MDPKO) by breeding DNA-PKcsfl/fl mice (Mishra et al., 2015) with mice expressing the Cre recombinase driven by muscle creatine kinase (MCK) (Figure 3I). As expected, in skeletal muscle, p-HSP90α was lower and p-AMPK and phosphorylation of AMPK substrate acetyl-CoA carboxylase (p-ACC) were higher in 17 mo old MDPKO mice compared to DNA-PKcsfl/fl (fl/fl) mice; there was less difference in 5 mo old mice. Taken together, these findings support the notion that the aging-related increase in DNA-PK-mediated phosphorylation of HSP90α leads to the decline of AMPK activity with age.
Since DNA-PK is involved in NHEJ, we asked whether DNA-PK-deficiency or treatment with DNA-PK inhibitor NU7441 may affect the DNA DSB burden in skeletal muscle. As shown in Figure S3, γ-H2AX in skeletal muscle of SCID and MDPKO mice were similar or slightly decreased (in old SCID) compared to WT mice; treatment with NU7441 also did not affect γ-H2AX levels.
DNA-PK inhibition increases skeletal muscle mitochondrial content
These findings led us to consider the possibility that DNA-PK may promote aging-associated mitochondrial decline in skeletal muscle. We quantified mitochondrial content in skeletal muscle of 14–16 year old rhesus monkeys and its correlation with DNA-PKcs activity. We found a strong inverse correlation between mitochondrial content and p-DNA-PK in middle aged skeletal muscle (r=−0.93, p=0.008) (Figure 4A). One possible interpretation of this finding is that DNA-PK suppresses mitochondrial biogenesis in middle aged skeletal muscle.
Figure 4. DNA-PK inhibition increases skeletal muscle mitochondrial biogenesis.
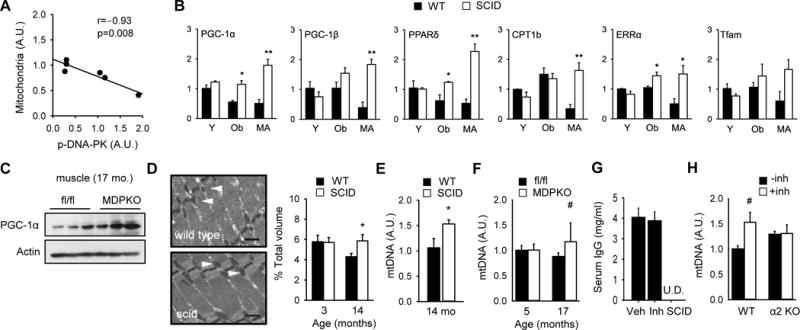
(A) Correlation between mitochondrial content and DNA-PKcs autophosphorylation in skeletal muscle of middle aged (14–16 yr) rhesus monkeys (r=−0.93, p=0.008). The correlation values were calculated by Pearson correlation. 95% confidence interval was −0.99 to −0.46. (B) Skeletal muscle was isolated from resting young (Y, 3–4 mo), obese (Ob, 5–6 mo) and middle aged (MA) WT and SCID mice. The mRNA levels (in arbitrary units) of genes important for mitochondrial biogenesis were measured by using real-time PCR (n=3–4 per group). All values are given as mean ± s.e.m. *, p<0.05, **, p<0.01 between WT and SCID values. (C) PGC-1α expression level in resting muscle isolated from old (17 months old) fl/fl and muscle specific DNA-PK knockout mice (MDPKO). (D) (Left) A representative electron micrograph of gastrocnemius isolated from middle aged WT and SCID mice (×10,000 magnification). Black bar indicates 500 nm. White arrowhead indicates mitochondria. (Right) Mitochondrial volume, expressed as percentage of total muscle volume, is shown for 3 mo and 14 mo WT and SCID mice. *, p<0.05 between WT and SCID values. (E) Relative mtDNA levels in skeletal muscle of middle aged WT and SCID mice (n=5–6 per genotype). *, p<0.05 between WT and SCID values. (F) Relative mtDNA levels in skeletal muscle of young (5 mo) and old (17 mo) fl/fl and MDPKO mice (n=5 per genotype). #, p=0.06. (G) Serum IgG levels of C57BL6/J mice treated with DNA-PK inhibitor for 10–12 week. Serum IgG was undetectable (U.D.) in SCID mice (n=5–13 per group). (H) Relative mtDNA levels in skeletal muscle of WT and AMPKα2 knockout mice fed either vehicle or DNA-PK inhibitor NU7441 (n=4–5 per genotype). #, p=0.06.
Peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α) (Puigserver et al., 1998) is the master regulator of mitochondrial function. We measured mRNA levels of PGC-1α as well as related PGC-1β and additional genes involved in mitochondrial function in skeletal muscle isolated from WT and SCID young mice (Y, 3–4 mo old), obese mice generated by feeding high-fat diet (HFD) for 4–5 months (Ob, 5–6 mo old) and middle aged mice (MA, 12–14 mo old) (Figure 4B). We found that mRNA levels of PGC-1α and PPARδ, were decreased in muscle from obese WT mice but not from SCID mice fed a HFD. Also, the expression of all the genes tested was decreased in middle aged WT muscle, but not in middle aged SCID muscle. Consistent with this, the protein levels of PGC-1α in skeletal muscle of 17 mo old MDPKO mice were elevated compared to the controls (Figure 4C).
We then calculated the mitochondrial density in skeletal muscle of young and middle aged WT and SCID mice. Morphometric analyses of electron micrographs indicated that middle aged WT muscle had 36% less mitochondrial density than young WT muscle (Figure 4D). However, middle aged SCID muscle had same mitochondrial density as young SCID and WT muscle. In agreement with this, the mtDNA copy number in middle aged muscle was higher in SCID mice (Figure 4E) and tended to be higher in MDPKO mice (Figure 4F) compared to their respective controls.
These findings imply that inhibition of DNA-PK activity with a small molecule will induce mitochondrial biogenesis. We treated WT mice with highly-specific DNA-PK inhibitor NU7441 for 3 mo by incorporating it in the HFD pellet, starting from age 1 mo. After treatment, we measured serum IgG levels to determine if the inhibitor caused any immune dysfunction. Unlike SCID mice, which do not produce any antibody because of the lymphocyte defect, WT mice treated with NU7441 had normal levels of serum antibodies (Figure 4G), indicating that in adult mice, the DNA-PK inhibitor did not significantly affect B-cell function. To determine the role of AMPK in mitochondrial biogenesis, we also treated AMPKα2 KO mice (Viollet et al., 2003) on HFD with the inhibitor. As shown in Figure 4H, NU7441 increased mitochondrial content by approximately 50% in WT mice but did not increase it in AMPKα2 KO mice. Taken together, these findings are consistent with the notion that inhibiting DNA-PK increases mitochondrial biogenesis in an AMPK-dependent manner.
DNA-PK inhibition increases physical fitness in obese and middle aged animals
Skeletal muscle is composed of oxidative and glycolytic fiber types. Type 1 and type 2A fibers are high in mitochondrial content and are oxidative whereas type 2X and type 2B fibers are lower in mitochondrial content and are glycolytic. Previous studies have shown that increased expression of PGC-1α causes fiber switch from glycolytic toward more oxidative fibers (Lin et al., 2002). We analyzed the fiber type composition of gastrocnemius muscle, which is largely glycolytic, from middle aged SCID and WT mice and WT mice treated with the DNA-PK inhibitor for 10–12 weeks. As shown in Figure 5A, SCID muscle had more Type 1 fiber and less Type 2B fiber than WT muscle as did the muscle from the inhibitor treated mice (Figure 5B). Consistent with increased mitochondrial content and oxidative fiber in middle aged SCID muscle, they had higher mitochondrial oxygen consumption rate (VO2) in permeabilized muscle (Figure S4A).
Figure 5. DNA-PK inhibition increases physical fitness in obese and middle aged animals.
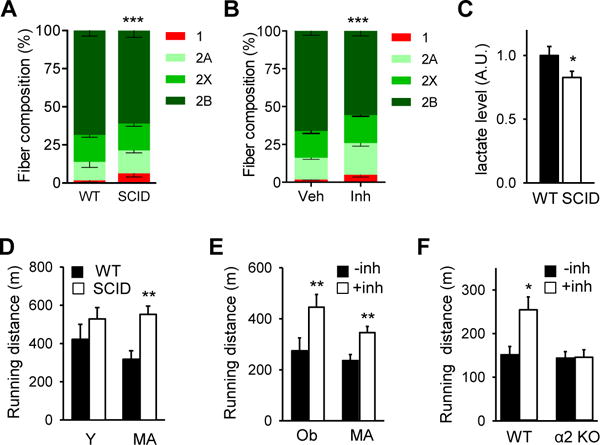
Gastrocnemius muscle fiber composition in middle aged WT and SCID mice (A) and in WT middle aged mice fed vehicle or the DNA-PK inhibitor for 10–12 wk (B) (n=5 per group). All values are given as mean ± s.e.m. ***, p<0.001 (Two-way ANOVA) between genotype and treatment groups. (C) Resting serum lactate levels in middle-age WT and SCID mice. Mann-Whitney Test: *, p<0.05 between WT and SCID values. (D) The distance (in meters) young (Y, 4 months old) and middle aged (MA, 12 mo) WT and SCID mice ran on the treadmill before exhaustion. For young, n=8; middle aged, n=8, for each genotype. Mann-Whitney Test: **, p<0.01 between WT and SCID values. (E) The distance middle aged (MA) and obese (Ob) mice treated with either vehicle or DNA-PKcs inhibitor ran on the treadmill before exhaustion (n=10 per treatment group). Mann-Whitney Test: **, p<0.01 between the treatment groups. (F) The distance obese WT and AMPKα2 knockout mice treated with either vehicle or DNA-PKcs inhibitor ran on the treadmill before exhaustion (n=4–6 per treatment group). Mann-Whitney Test: *, p<0.05 between the treatment groups.
Treatments that activate AMPK and/or reprogram glycolytic fibers, which are prone to fatigue, to oxidative fibers are known to increase fat oxidation and fatigue-resistance (Narkar et al., 2008; Suwa et al., 2003; Wang et al., 2004). Mice with increased mitochondrial content in skeletal muscle generally have lower serum lactate, the by-product of glycolysis and a major source of muscle fatigue (Hanson and Hakimi, 2008; Mason et al., 2004). Since SCID muscle has more oxidative fibers, we suspected that SCID mice might have lower serum lactate levels. Indeed, serum lactate levels of middle aged SCID mice were 17% lower than those of WT mice (Figure 5C) in the resting state.
These findings suggest that DNA-PK may promote aging-associated decline in physical fitness. To test this, we measured the treadmill running capacity of young and middle aged WT and SCID mice. Young WT and SCID mice ran similar distances, but middle aged SCID mice ran nearly twice the distance of middle aged WT mice, showing no aging-associated decline (Figure 5D). With some high-endurance mice, repeated exercise damages muscle and decreases endurance (Mason et al., 2004). This was a potential concern because increased mitochondrial content could lead to increased oxidative damage, including DNA damage. To test whether this is the case with SCID mice, we exercised obese and middle aged WT and SCID mice for three consecutive days (Figure S4B). By the third day of exercise, the superior endurance of SCID mice compared to WT mice became even more pronounced, indicating that SCID muscle is not prone to damage by repeated exercise.
Results shown above suggest that the DNA-PK inhibitor may be an effective fitness enhancer. Indeed, obese and middle aged WT mice treated with NU7441 ran approximately 60% and 40% greater distance, respectively (Figure 5E). Consistent with our earlier finding that DNA-PK inhibition does not increase mitochondrial biogenesis in skeletal muscle of obese AMPKα2 mice (Figure 4H), the inhibitor increased the running distance by more than 60% in obese WT mice but had no effect on the running distance in obese AMPKα2 KO mice (Figure 5F). Taken together, these findings indicate that DNA-PK inhibition can prevent aging-associated loss of mitochondria and its function and dramatically improve fitness in an AMPK-dependent manner.
To determine if the immune status of SCID plays any role in their physical performance, we exercised mice deficient in Rag1, a nuclease that is essential for VDJ recombination and lymphocyte development (Oettinger et al., 1990). Since Rag1 and DNA-PKcs participate sequentially in the same VDJ recombination pathway in lymphocytes, the immune status of Rag1 KO and SCID mice are very similar. Unlike SCID muscle, Rag1 KO muscle did not express higher PGC-1α or PPARδ expression (Figure S5A) and had normal physical endurance (Figure S5B).
Inhibition of DNA-PK protects against obesity and type-2 diabetes
To investigate the metabolic function of DNA-PKcs in vivo, we measured the body weight of young WT and SCID mice fed either regular chow diet (RCD) or HFD after weaning. After 20 wk on RCD, SCID mice weighed 1–2 gm less than WT mice, but the difference was statistically not significant (Figure S6A). On HFD, SCID mice were relatively resistant to weight gain (Figure S6B) compared to WT mice even though their food intake (Figure S6C) and fat absorption (Figure S6D) were similar to those of WT mice. NMR spectroscopy analysis indicated that on HFD, SCID mice have approximately 25% lower fat mass index than WT mice (Figure S6E). Consistent with this, fat in the abdominal region was noticeably decreased in SCID mice (Figure S6F and G). On the other hand, WT and SCID mice had similar lean mass index (Figure S6H).
Although the body weight difference between WT and SCID mice was insignificant at young age (see Figure S6A), it became much more pronounced at middle aged (14 mo) (Figure S6I). Consistent with this, middle aged SCID mice had lower plasma triglyceride levels than WT mice (Figure S6J). Middle aged SCID mice had similar fasting plasma glucose levels as WT mice (92 ± 8 mg/dl vs 94 ± 14 mg/dl) despite having significantly lower plasma insulin levels than WT mice (0.28 ± 0.01 ng/ml vs 0.64 ± 0.1 ng/ml, p<0.05). This suggested that middle aged SCID mice are less insulin resistant than WT mice. Indeed, the insulin resistance index (HOMA-IR) increased with age in WT mice as expected but did not change in SCID mice (Figure S6K). Taken together, these results indicate that DNA-PK promotes aging-associated increase in insulin resistance.
The results shown above suggested that a DNA-PK inhibitor might have an anti-diabetic effect in obese WT mice. Indeed, we found that including NU7441 in the pellet decreased weight gain on HFD (Figure 6A) and increased glucose tolerance (Figure 6B) and insulin sensitivity (Figure 6C). Activation of AMPK can increase the secretion of glucagon-like peptide-1 (GLP-1), a peptide hormone produced by the L-cells in the gut that helps to maintain normoglycemia (Baggio and Drucker, 2007). Consistent with the DNA-PK-AMPK link, the inhibitor increased the plasma level of GLP-1 (Figure 6D). To determine the site of improved insulin action in these mice, we performed hyperinsulinemic-euglycemic clamps. The glucose infusion rate (GIR) required to maintain euglycemia was approximately 27% higher in inhibitor-treated mice (Figure 6E, Figure S7A, B) and the total glucose disposal rate (GDR) was increased by almost 20% (Figure 6F). The inhibitor increased insulin-stimulated glucose uptake (GU) into skeletal muscle, the primary site of insulin-stimulated glucose disposal, by almost 40% (Figure 6G) and insulin-stimulated glucose uptake into white adipose tissue (WAT) by more than 120% (Figure 6H). However, endogenous glucose production (EGP) by the liver was not improved by NU7441 (Figure 6I). Middle aged MDPKO mice also tended to have lower body weight, fasting glucose and insulin resistance compared to fl/fl mice although the differences were not statistically significant (Figure S7C). These findings indicate that inhibition of DNA-PK activity with a small molecule inhibitor protects against obesity and has an anti-diabetic effect.
Figure 6. Inhibition of DNA-PK protects against obesity and type-2 diabetes.
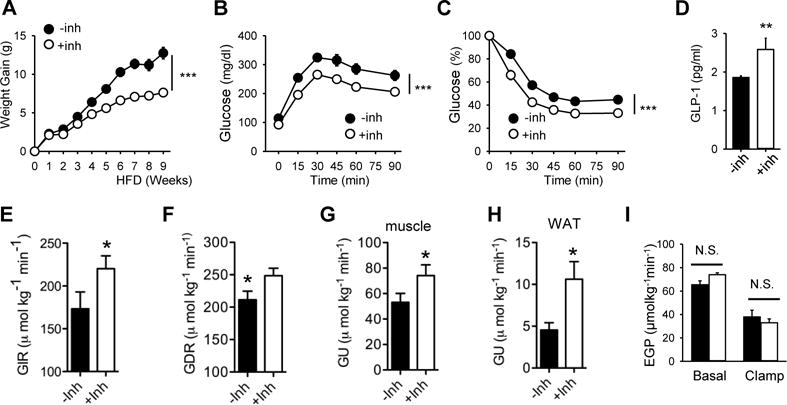
(A) Weight gain of WT mice fed HFD (±NU7441) (n=10 per treatment group). All values are expressed as mean ± s.e.m. ***, p<0.001 (Two-way repeated measures ANOVA) between the treatment groups. Glucose tolerance (B) and insulin sensitivity (C) of mice on HFD (±NU7441) for 10–12 wk are shown (n=10 per treatment group). ***, p<0.001 (Two-way repeated measures ANOVA) between the treatment groups. (D) Plasma concentrations of GLP-1 in mice fed HFD (±NU7441) (n=7–8 per treatment group). Mann-Whitney Test: **, p<0.01 between the treatment groups. A hyperinsulinemic-euglycemic clamp study was performed on mice fed HFD (±NU7441) (n=7–8 per treatment group) to measure (E) glucose infusion rate (GIR), (F) glucose disposal rate (GDR), (G and H) glucose uptake (GU) in skeletal muscle (gastrocnemius) and WAT, respectively, and (I) endogenous glucose production (EGP) from the liver. N.S., not significant; *, p<0.05 between the treatment groups.
Calorie restriction and aerobic fitness are associated with decreased DNA-PK activity
Calorie-restriction (CR), like DNA-PK inhibition, increases mitochondrial biogenesis (Lopez-Lluch et al., 2006) and protects against insulin resistance and metabolic syndrome (Omodei and Fontana, 2011). We examined how CR affected DNA-PK activity and phosphorylation of T5,7 in HSP90α in middle aged rhesus macaques. CR, 30% caloric reduction compared to ad libitum feeding for 3.4 yr, decreased both DNA-PKcs autophosphorylation and HSP90α phosphorylation in middle aged skeletal muscle (Figure 7A).
Figure 7. Calorie restriction and aerobic fitness are associated with decreased DNA-PK activity.
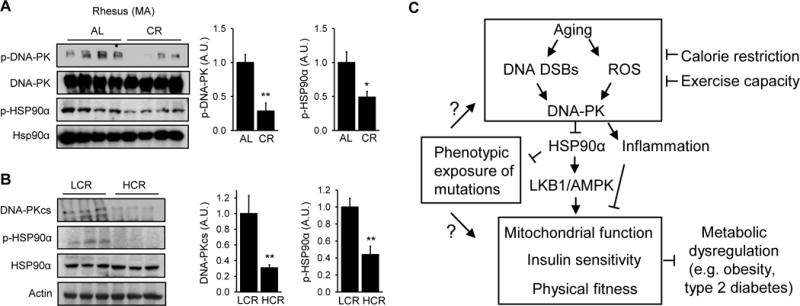
(A) (Left) DNA-PKcs autophosphorylation and p-HSP90α in skeletal muscle of ad libitum (AL) fed and calorie-restricted (CR, for 3.4 yr) middle aged rhesus macaques (n=4 per group). (Right) Quantification of DNA-PKcs autophosphorylation and p-HSP90α. All values are expressed as mean ± s.e.m. Mann-Whitney Test: *, p<0.05; **, p<0.01. (B) (Left) Total DNA-PKcs level and p-HSP90α in skeletal muscle of LCR (Low Capacity Runners), HCR (High Capacity Runners) rats. (Right) Quantification of DNA-PKcs expression level and p-HSP90α in skeletal muscle of LCR and HCR rats (n=10 per group). Mann-Whitney Test: **, p<0.01. (C) Schematic diagram showing how DNA-PK links aging-associated DNA DSBs to diminished AMPK activity and metabolic functions in skeletal muscle.
A number of clinical studies during the past two decades have shown that low exercise capacity is a strong predictor of morbidity and mortality (Kokkinos et al., 2008). As part of an effort to understand the link between exercise capacity and health, rats with low and high intrinsic treadmill running capacity have been produced by two-way selective breeding of genetically heterogeneous founder rats (Koch et al., 2012). The lines, termed the Low Capacity Runners (LCR) and the High Capacity Runners (HCR), differed by approximately 7-fold in running capacity after 27 generations of selective breeding. The sedentary HCR rats have increased mitochondrial content and function, are protected from the metabolic syndrome, hypertension, type 2 diabetes and cancer and have longer lifespan compared to the sedentary LCR rats (Garton et al., 2016). Since the HCR rats have metabolic phenotypes that resemble CR and DNA-PK-deficiency, we hypothesized that breeding for the HCR rats may have led to the generation of rats with decreased DNA-PK activity. We measured total DNA-PKcs level and p-HSP90α in skeletal muscle of 7–9 mo sedentary the HCR and LCR rats. As shown in Figure 7B, DNA-PKcs level was three-fold lower and p-HSP90α was approximately 60% lower in the HCR rats compared to LCR rats. Taken together, these findings suggest that CR and aerobic fitness, which delay aging and protect against chronic diseases, are associated with decreased DNA-PK activity.
DISCUSSION
In western societies, approximately 30% of the population becomes obese by age 60–70 and 40% becomes either prediabetic or diabetic by age 70–80 (Harris, 1993). Moreover, the capacity for physical exercise, which has protective effects against obesity as well as many diseases of aging, also declines with age. Obesity and aging are also dominant risk factors for many common diseases including type 2 diabetes, coronary artery disease, hypertension, stroke, cancer and even Alzheimer’s disease. Since the percentage of people age 65 or more is rapidly increasing, understanding how aging negatively affects energy metabolism and exercise capacity is imperative.
Cells respond to conditions in which energy demand exceeds respiratory capacity by expanding mitochondrial content, but with increasing age, mitochondrial content and function declines. One clue to the mechanism for their decline was the discovery that aging impairs AMPK-induced mitochondrial biogenesis in skeletal muscle (Lee et al., 2010; Reznick et al., 2007) although the reason for this was not known. Our findings indicate that aging increases DNA DSBs and DNA-PK-mediated phosphorylation of HSP90α, which results in decreased chaperone function for LKB1/AMPK (Figure 7C).
The long-held belief has been that aging-associated forms of deterioration, including the mitochondrial decline in skeletal muscle, are due to passive accumulation of damage to DNA, proteins and/or lipids with age. While these accumulated damages may contribute to aging-associated metabolic decline, our findings indicate that a genetic program activated by DNA-PK (Figure 7C) contributes to the metabolic and physical decline at older age. Aging is also associated with inflammation, which is a common denominator of most chronic diseases, including type 2 diabetes (Lumeng and Saltiel, 2011). Therefore, it is possible that the proinflammatory effects of DNA-PK (Mishra et al., 2015) may also contribute to the metabolic dysfunction associated with obesity and aging.
Although both DNA DSBs and DNA-PK activity increase with aging in skeletal muscle (Figure 2A), we cannot conclude that aging-associated increase in DNA-PK activity is solely mediated by chromosomal breaks as DNA-PK is also activated independently of chromosomal breaks by ROS (Li et al., 2014), which increase with aging (Lee et al., 2010). In light of our finding that DNA-PK activity decreases mitochondrial function in skeletal muscle of older mice, activation of DNA-PK by ROS is an intriguing possibility as mitochondria are the main source of ROS production in metabolic tissues and CR decreases ROS (Sohal and Weindruch, 1996).
The most prominent phenotype of SCID mice is lymphocyte deficiency. However, mice deficient in Rag1, which functions in the same pathway as DNA-PK in VDJ rearrangement in lymphocytes (Oettinger et al., 1990) had similar mitochondrial gene expressions and physical fitness (Figure S5). Consistent with this, Liu et al. (Liu et al., 2015) reported that Rag1−/− mice are not leaner than WT mice on high-fat diet. Taken together, these findings indicate that the lymphocyte deficiency does not play a significant role in the metabolic phenotype of SCID mice. Nevertheless, we acknowledge that some of the changes seen in SCID tissues may be due to tissue cross-talk. For example, we cannot rule out the possibility that some phenotype in SCID skeletal muscle may have been caused by decreased adiposity.
Our finding that SCID mutation or treatment with DNA-PK inhibitor protects against aging-associated decline in metabolism and physical fitness is at odds with the observation that DNA-PK−/− mice undergo accelerated aging (Espejel et al., 2004). The most likely explanation for this paradox is that the SCID mutation is leaky and the DNA-PK inhibitor does not inhibit DNA-PK completely. The residual DNA-PK activity may have some protective function against the level of DNA DSBs generated naturally. Moreover, in the non-lymphoid cells, the Mre11-Rad50-Nbs1 complex may contribute to NHEJ (Rass et al., 2009) in a redundant manner.
In addition to the increase in DNA DSBs, another change that occurs with aging is the decline of NAD+, the cofactor for Sirt1 (Imai and Guarente, 2014). As Sirt1 plays an important role in mitochondrial function and energy metabolism, the decline in NAD+ levels may contribute to mitochondrial and metabolic decline. The roles of Sirt1 and DNA-PK in aging-related changes may be interlinked as AMPK promotes the synthesis of NAD+ by regulating the NAD+ biosynthetic enzyme Nampt (Fulco et al., 2008).
The biochemical function of HSP90 is to fold metastable proteins. One of the important concepts to come out of the HSP90 field is that it is a capacitor of phenotypic variation (Queitsch et al., 2002). That is, by acting as a buffer, HSP90 allows mutations or polymorphisms (inherited or acquired) to accumulate unseen, free from the pressures of natural selection. As demand for HSP90 function increases with stress, HSP90 is no longer able to shield these mutations, leading to a diverse array of defects. Adopting this concept to our work, HSP90α T5,7 phosphorylation, combined with the aging-associated stress, might decrease the ability of HSP90α to provide the necessary buffering at older age, at least in skeletal muscle (Figure 7C). As a consequence, chaperone function may be decreased and mutations or polymorphisms may be exposed at older age. In this scenario, aging and aging-associated diseases may partly be a manifestation of the exposure of these mutations or polymorphisms.
Weight gain and lack of exercise is often attributed to poor lifestyle choices. One’s lifestyle is ultimately the individual’s choice, but we must acknowledge that strong underlying forces that cause metabolic and physical decline make healthy lifestyle goals difficult to attain with aging. Understanding the molecular basis of these forces will help us address more effectively the public health challenges brought on by obesity, lack of exercise and aging.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Jay H. Chung (chungj@nhlbi.nih.gov)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture and siRNA transfection
C2C12 cells were obtained from American Type Culture Collection (ATCC) and grown in complete Dulbecco’s modified Eagle’s medium (DMEM) medium supplemented with 10% fetal bovine serum (FBS) (growth media) and 100 g/ml penicillin and streptomycin. C2C12 cells were used before passage 25. When C2C12 were nearly confluent (70%–80% confluency), they were induced to differentiate by replacing the growth media with the differentiation media (DMEM supplemented with 2% horse serum). For studies that use dexamethasone, we used charcoal-stripped 2% horse serum (Invitrogen) in the differentiation media. The mouse DNA-PKcs (ON-TARGETplus Mouse Prkdc (19090) siRNA - SMARTpool), HSP90α (ON-TARGETplus Mouse Hsp90aa1 (15519) siRNA - SMARTpool, 5 nmol) were purchased from Dharmacon. Control siRNA (ON-TARGETplus Non-targeting siRNA #1) was also from Dharmacon. DNA-PKcs or HSP90α was knocked down by transfecting 80–90% confluent C2C12 cells with siRNA by using Lipofectamine 2000 according to the manufacturer’s protocol. Cells were switched to low serum media (2% horse serum) to induce differentiation. Three days after transfection, cells were harvested and lysed.
Mice and rats
All experiments were approved by the ACUC (Animal Care and Use Committee) of the NHLBI and the University of Utah. Male C57BL/6, PrkdcSCID (B6.CB17-PrkdcSCID), B6.129S7-Rag1tm1Mom/J and B6.FVB(129S4)-Tg(Ckmm-cre)5/Khn mice were purchased from The Jackson Laboratories (Bar Harbor, Maine). DNA-PKcsfl/fl mice(Mishra et al., 2015) were bred with B6.FVB(129S4)-Tg(Ckmm-cre)5/Khn mice to generate DNA-PKcsfl/fl; CKmm-Cre (MDPKO) mice that were homozygous at each allele. AMPKα2KO mice (Viollet et al., 2003) were backcrossed to C57BL/6J for at least six generations. Unless indicated otherwise, mice were fed Lab Diet 5021. We used male littermate controls for all mouse studies.
For DNA-PK inhibitor (NU7441) studies in middle-age mice, mice were dosed once daily by oral gavage with or without 40 mg/kg/d NU-7441 in 10% PEG400 in saline for up to 12 weeks. 10 l/g body weight was dosed by oral gavage. For high fat diet (HFD) studies with DNA-PK inhibitor in WT and/or AMPKα2KO mice, NU7441 was mixed with HFD (40% fat by calories, D12327, Research diet, New Brunswick, USA) at a concentration of 3g/kg of food to provide a 300 mg/kg dose, and pellets were then reconstituted. Control groups received pellets without NU7441. Body weight and caloric intake were monitored throughout the experiments. Mice were housed with a 12 hr light-dark cycle (light on 6 am–6 pm) with free access to food and water.
To induce obesity in WT, SCID and RAG1−/− mice, they were fed HFD (60% fat by calories, D12492, Research Diets) for 5 mo starting immediately after weaning (3–4 weeks of age). Regular chow diet (RCD, 12% fat by Kcal) was purchased from Zeigler (Rodent NIH-31). Body weight was recorded monthly and food intake was measured every second day for 14 consecutive days.
The LCR and HCR rats were birthed and subsequently tested for treadmill endurance running capacity when 11 weeks old using procedures on an IACUC protocol approved by the University of Michigan. Only Male Rats were shipped from Ann Arbor, Michigan, via air courier at 6 mo age. Tissues were harvested from these rats at 7–9 mo age.
Rhesus Macaque tissue analysis
Animal care was provided in accordance with the NIH Guide for the Care and Use of Laboratory Animals and this research was approved by the Institutional Animal Care and Use Committee of the Oregon National Primate Research Center. Male rhesus macques (Macaca mulatta) ranging in age 1–16 y.o. were used for the aging studies. After overnight fasting, animals were anesthetized and gastrocnemius and subcutaneous fat were biopsied. Female rhesus macaques (10–12 y.o.) were matched by body weight and age, then assigned to ad libitum control or 30% calorically-restricted (CR) treatment groups for 3.4 y. The monkeys were singly-caged indoors at a temperature of 24°C under a fixed 12 h light:12 h dark photoperiod, with unlimited access to drinking water. All animals received a specially formulated monkey chow that included additional vitamin and minerals to avoid any deficiencies in essential nutrients. Feedings were conducted at 0800 h and 1500 h each day, but with CR animals receiving 30% fewer calories, as previously described (Ingram et al., 1990; Ingram et al., 1993; Mattison et al., 2003). Monkeys were also supplemented with daily fresh fruits or vegetables.
| Ingredient | Amount (% by Weight) | |
|---|---|---|
| Basal Mix | ||
|
| ||
| Ground wheat | 35.5 % | |
| Ground corn | 22.1 % | |
| Soybean hulls | 12 % | |
| Soybean meal (48% protein) | 8.5 % | |
| Fish meal (60% protein) | 5.47 % | |
| Sugar | 4 % | |
| Alfalfa meal | 3 % | |
| Dried whey | 3 % | |
| Brewers yeast | 2 % | |
| Limestone | 1.3 % | |
| Dicalcium phosphate | 1 % | |
| Iodized salt | 0.6 % | |
| Mineral mix | 0.4 % | |
| dl-methionine | 0.13 % | |
| Vitamin mix | 1 % | |
|
| ||
| Complete Diet | ||
|
| ||
| Basal mix | 96.75 % | |
| Soybean oil | 3.1 % | |
| Vitamin C | 0.15 % | |
|
| ||
| Mineral Premix | ||
|
| ||
| Mineral | Amount (per 100 lb product) | Source |
|
| ||
| Cobalt | 30 mg | Cobalt carbonate |
| Copper | 500 mg | Copper sulfate |
| Iron | 3 g | Iron sulfate |
| Magnesium | 22 g | Magnesium oxide |
| Manganese | 3 g | Manganous oxide |
| Potassium | 25 g | Potassium bicarbonate |
| Zinc | 3.7 g | Zinc oxide |
| Iodine | 100 mg | Calcium iodate |
|
| ||
| Vitamin Premix | ||
|
| ||
| Vitamin | Amount (per 100lb product) | Source |
|
| ||
| Vitamin A | 680,000 IU | Stabilized Vitamin A |
| Vitamin D | 230,000 IU | D-3 |
| Vitamin E | 3 g | dl-alpha-tocopheryl acetate |
| Vitamin B12 | 850 mcg | |
| Riboflavin | 350 mg | |
| Niacin | 3 g | |
| Pantothenic acid | 3 g | d-calcium pantothenate |
| Choline | 20 g | Choline chloride |
| Meandione activity | 500 mg | |
| Folic acid | 400 mg | |
| Thiamin | 250 mg | Thiamin mononitrate |
| Pyridoxine | 500 mg | |
| Biotin | 5 mg | d-biotin |
METHODS DETAILS
Body composition analysis
Body fat and lean mass were measured by NMR (Bruker BioSpin Corporation, Houston, TX). Body fat indices were calculated by dividing fat or lean mass by body weight.
Insulin tolerance test (ITT) and Glucose tolerance test (GTT)
Plasma glucose was measured by using a glucometer (Ascensia). For the glucose tolerance test and insulin tolerance test, mice were fasted for 16 hr, and 1 mg/g glucose or 0.75 mIU/g insulin were injected intraperitoneally (i.p.). Blood glucose was measured at 0, 15, 30, 45, 60 and 90 min after injection.
Treadmill endurance test
Prior to the treadmill endurance tests, the mice were trained by running on an Exer-3/6 mouse treadmill (Columbus Instruments) at 10 m/min for 5 minutes for 2 days. For the endurance test, the treadmill was set at a 15° incline, and the speed was increased in a stepwise-fashion (10 m/min for 10 min followed by 14 m/min for 5 min and then the final speed of 18 m/min). The test was terminated when mice reached exhaustion, which was defined as immobility for more than 30 sec.
Fat absorption
A synthetic diet containing 5% sucrose polybehenate, which is not absorbed, was fed to WT and SCID mice for 3 days. Fecal samples were collected and analyzed for fatty acid methyl esters by gas chromatography. Fat absorption was calculated from the ratio of behenic acid to the other fatty acids in the diet.
Serum analysis
Serum nonesterified fatty acid and triglyceride were measured by spectrophotometric enzymatic assay (Free fatty acids, half micro test; Roche Diagnostics and Infinity Triglycerides liquid Stable Reagent; Thermo Scientific). Serum GLP-1 levels were measured using GLP-1 (active) ELISA (Millipore), according to the manufacturer’s directions. Serum insulin level was determined using an ELISA kit (EMD Millipore). Serum lactate level was measured by colorimetric assay kit (Eton Bioscience), serum IgG level was measured by easy-titer mouse IgG assay kit (Thermo Scientific).
Real-time PCR
Frozen skeletal muscles, liver, WAT and BAT were ground and homogenized in a mortar and pestle under liquid nitrogen. Total RNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s protocol. Complementary DNA (cDNA) was synthesized from 2 μg of DNA-free total RNA using High Capacity cDNA Archive kit (Applied Biosystems). 10 μl of RNA (2 g) was mixed with 10 μl reverse transcriptase master mix (2X), which contained 2 μl 10× RT buffer, 2 μl 10X Random Primers, 0.8 μl 25X dNTP mix, 1 μl MultiScribe RT (50 U/ul) and 4.2 μl RNAse free water, giving a final volume of 20 μl. The reaction mixtures were incubated at 25°C for 10 min and 37°C for 120 min. Reverse transcription products were diluted 1:3 in nuclease-free water, and 1 μl was used for RT-PCR with the Taqman core reagents RT-PCR kit (Applied Biosystems) in combination with the 7900T Real-Time PCR System (Applied Biosystems). The RT-PCR was performed for 40 cycles at the following cycling condition: 5°C for 10 min initial denaturation, then 40 cycles at 95°C denaturation, 60°C anneal/extension for 15 sec and 1 min at each temperature, respectively. 18S RNA was used as the internal standard for all mRNA. The following primers (Applied Biosystems) were used in these studies: PGC-1α, Mm00447183_m1; PGC-1β, Mm00504720_m1; PPARδ, Mm00803186_g1; CPT1b, Mm00487200_m1; ERRα, Mm00433143_m1; Tfam, Mm00447485_m1; Euk 18SrRNA, 4333760F; AMPKα, Mm01264789_m1.
Transmission Electron Microscopy
The samples were fixed for 1 h in a mixture of 2.5% glutaraldehyde, 4% paraformaldehyde, in phosphate buffer (pH 7.4), washed in distilled water, and placed in 1% osmium for 1 hour. The samples were then washed again and dehydrated with acetone before infiltration and embedding with EPON 812. The EPON-embedded samples were baked at 60°C for 48 h. Ultrathin sections (about 60–90nm) were cut on a Leica Ultracut Ultramicrotome, picked up on to copper grids stained with uranylacetate and lead citrate, and examined in a JEOL 1200EX Transmission Electron Micoscope (JEOL).
Immunoblotting and reagents
Cells were lysed in RIPA buffer and subjected to immunoblotting. The following antibodies were used: ACC1 (Cell Signaling); p-ACC1 (Cell ignaling); AMPK (Cell Signaling); p-AMPK(T172) (Cell Signaling); H2AX (Cell Signaling); phospho-H2AX(S139) (Cell Signaling); DNA-PK (Lab Vision); p-DNA-PK(S2056) (Abcam); LKB1 (Santa Cruz); HSP90α (Millipore); p-HSP90(T5,7) (Cell Signaling); PGC-1α (Santa Cruz); Cyto C (Cell Signaling); Actin (Santa Cruz). Densitometry was performed with the ImageJ software (NIH, Bethesda, MD, USA).
Whole-cell lysate preparation
Cell pellets were lysed on ice for 20 min in RIPA buffer (50 mM Tris-HCl, pH 7.4, 0.15 M NaCl, 1.0 mM EDTA, 1% NP-40, 0.25% sodium deoxycholate) freshly supplemented with phosphatase and protease inhibitors (Millipore). Lysates were clarified by centrifugation at 14,000 rpm for 10 min. Proteins were quantified using the Coomassie plus protein.
Mitochondrial DNA (mtDNA) Quantification by Quantitative Real-Time PCR
Relative amounts of nuclear DNA and mtDNA were determined by quantitative Real-Time PCR. The ratio of mtDNA to nuclear DNA reflects the tissue concentration of mitochondria per cell. Skeletal muscle tissue were homogenized and digested with Proteinase K overnight in a lysis buffer for DNA extraction by DNeasy blood and tissue kit (QIAGEN). Quantitative PCR was performed using each primers (mtDNA specific PCR, forward 5′-CCGCAAGGGAAAGATGAAAGA-3′, reverse 5′-TCGTTTGGTTTCGGGGTTTC-3′; and nuclear DNA specific PCR, forward 5′-GCCAGCCTCTCCTGATTTTAGTGT-3′, reverse 5′-GGGAACACAAAAGACCTCTTCTGG-3′) and Power SYBR Green PCR master mix (Applied Biosystems) in a 7900T Real-Time PCR system (Applied Biosystems). The PCR reactions consisted of 10 μl Power SYBR Green PCR Master mix (2X), 7 μl RNAse free water, 1 μl 300 nM primer mix and 2 μl cDNA, to a total volume of 20 μl. Three technical replicates were performed for each sample. The cycling conditions were 15 minutes at 95° C, followed by 50 to 60 cycles of 15 seconds at 95° C, 20 seconds at 58° C and 20 seconds at 72° C as previously described(Lagouge et al., 2006).
Tissue lysate preparation
Frozen tissues were ground and homogenized in a mortar and pestle under liquid nitrogen. Samples were then incubated in RIPA buffer (50 mM Tris-HCl, pH 7.4, 0.15 M NaCl, 1.0 mM EDTA, 1% NP-40, 0.25% sodium deoxycholate) freshly supplemented with phosphatase and protease inhibitors (Millipore). In order to break the tissue up further and to shear DNA, lysates were sonicated briefly (5 sec, 140 watt, setting 7 using Ultrasonics W-385 Sonicator (Heat Systems), which were then vortexed every 30 minutes. The homogenates were incubated at 4°C for 2 h and then centrifuged (13,000 rpm) for 15 min at 4°C, and supernatants were collected.
Muscle fiber analysis
Muscle tissue lysates were prepared in a sample buffer consisting of dithiothreitol (DTT, 100 mM), sodium dodecyl ulphate (SDS, 2%), Tris-base (80 mM) pH 6.8, glycerol (10%) and Bromphenol Blue (1.2% w/v). Samples were boiled (2 min) and electrophoresed in SDS-PAGE. Total acrylamide concentration was 4% and 8% in the stacking and resolving gels, respectively (bis-acrylamide, 1:50). Gels (16′ 22 cm, 0.75 mm thick) were run at a constant current of 10 mA until voltage rose to 275 V, and thereafter at constant voltage for 16 h at 4±6°C. For each muscle a volume corresponding to 1.25 lg of total protein was loaded into a well and gels were silver-stained (BioRad, Hercules, CA). Myosin heavy chain bands were analyzed by using a BioRad GS-800 Calibrated Densitometer and quantified by usingQuantity One.
Morphometric analysis
Electron microscopic morphometric analysis of the volume density of mitochondria was carried out in young and middle aged mice. Micrographs of randomly selected areas of muscle fibers were obtained and scanned. These micrographs were analyzed with an image analysis software (Aperio imagescope).
Euglycemic-hyperinsulinemic clamp
The clamp studies were performed in conscious, restrained mice. Four days before the clamp experiment, mice were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine. A catheter was inserted into a lateral incision on the right side of the neck and advanced into the superior vena cava via the right internal jugular vein. The catheter was then sutured into place according to the protocol of MacLeod and Shapiro (MacLeod and Shapiro, 1988). Experiments were started at 0800 hours, after a 14-hour fast. The basal rates of glucose turnover were measured by continuous infusion of [3-3H]glucose (0.05 μCi/min) for 120 min, which followed a bolus of 2 μCi. Blood samples (20 μl) were taken at 90 and 115 min of the basal period for the determination of plasma [3H] glucose concentration. A 120-min hyperinsulinemic euglycemic clamp was started at 10:00 AM. Insulin was infused as a bolus of 18 mU/kg over a period of 3 min followed by continuous insulin infusion at the rate of 2.5 mU·kg−1·min−1 (Humulin R; Eli Lilly, Indianapolis, IN) to raise the plasma insulin concentration to ~1.5 ng/ml. During the clamp study, blood samples (20 μl) were collected via a small nick in the tail vein at 15-min intervals for the immediate measurement of plasma glucose concentration, and 20% glucose was infused at variable rates to maintain blood glucose at ~140 mg/dl in WT mice. Insulin stimulated whole body glucose flux was estimated using a continuous infusion of high-pressure liquid chromatography-purified [3-3H]glucose (0.1 μCi/min; NEN Life Science Products, Boston, MA) throughout the clamps. To estimate insulin-stimulated glucose transport activity and metabolism in skeletal muscle, 2-deoxy-D-[1-14C]glucose (NEN Life Science Products) was administered as a bolus (10 μCi) at 45 min before the end of clamps. Blood samples (20 μl) were taken at 80, 85, 90, 100, 110, and 120 min after the start of clamps for the determination of plasma [3H]glucose, 2-deoxy-D-[1-14C]glucose, and 3H2O concentrations. Additional blood samples (10 μl) were collected before the start and at the end of clamp studies for measurements of plasma insulin concentration. All infusions were performed using microdialysis pumps (CMA/Microdialysis, Acton, MA). At the end of the clamp period, animals were anesthetized with ketamine-xylazine injection. Within 5 min, gastrocnemius muscle from hindlimbs, epididymal and brown adipose tissue, and liver were removed. Each tissue, once exposed, was dissected within 2 s, frozen immediately using liquid nitrogen-cooled aluminum blocks, and stored at –70°C for later analysis. The determination of plasma [3-3H]glucose and 2-deoxy-D-[1-14C] glucose concentrations and tissue 2-deoxy-D-[1-14C] glucose-6-phosphate were performed as described previously (Youn et al., 1994). Basal endogenous glucose production was calculated as the ratio of the preclamp [3-3H]glucose infusion rate (dpm/min) to the specific activity of the plasma glucose (mean of the values in the 90 and 115 min of basal preclamp period, in dpm/μmol). Clamp whole-body glucose uptake was calculated as the ratio of the [3-3H]glucose infusion rate (dpm/min) to the specific activity of plasma glucose (dpm/μmol) during the last 30 min of the clamp (mean of the 90–120 min samples). Clamp endogenous glucose production was determined by subtracting the average glucose infusion rate in the last 30 min of clamp from the whole-body glucose uptake. Muscle and white and brown adipose tissue glucose uptake was calculated from the plasma 2-deoxy-D-[1-14C] glucose concentration profile (using plasma 14C counts at 80–120 min, the area under the curve was calculated by trapezoidal approximation) and tissue 2-deoxy-D-[1-14C] glucose-6-phosphate content as described previously (Youn et al., 1994). Tissue glycogen synthesis was calculated from 3H incorporation to glycogen (Toyoshima et al., 2005).
Mitochondrial function
Mitochondrial function was studied using the saponin-permeabilized fiber technique(Boudina et al., 2005; Boudina et al., 2007) on soleus muscle. Respirations were performed in 1mL KCl buffer (KCl 125mM, HEPES 20mM, Magnesium-Acetate 3mM, EGTA 0.4mM, KH2PO4 5mM, DTT 0.3mM, BSA 2mg/ml). State 2 respiration was measured after addition of 5 mM Succinate and 10 mM Rotenone (Complex I inhibitor). State 3 respiration was assessed by adding 1 mM ADP and state 4 respiration measured after addition of 1 μg/mL oligomycin (ATP synthase inhibitor).
HOMA-IR
The HOMA-IR formula and HOMA2-IR online calculator downloaded from https://www.dtu.ox.ac.uk/homacalculator/ were used to calculate HOMA-IR.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical comparisons between groups were analyzed by Two-way ANOVA and Two-way repeated measures ANOVA for a parametric test or Mann-Whitney test for a nonparametric test using Prism7. Results are expressed as the mean±s.e.m. Significance was accepted at p<0.05. The Excel software (Microsoft) was used to determine average values, standard errors and standard deviations. For each figure, number of experimental replicates and other information relevant for assessing the accuracy and precision of the analysis are included in the accompanying legend.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program, National Heart Lung and Blood Institute, in the National Institutes of Health. E. Dale Abel was supported by NIH grant HL73167. The LCR-HCR rat model system was funded by the Office of Research Infrastructure Programs grant P40OD021331 (to LGK and SLB) from the NIH. This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number : HI14C1176). We acknowledge the expert care of the rat colony provided by Molly Kalahar and Lori Heckenkamp. Contact LGK lgkoch@umich.edu or SLB brittons@umich.edu for information on the LCR and HCR rats: these rat models are maintained as an international resource with support from the Department of Anesthesiology at the University of Michigan, Ann Arbor, Michigan. We thank Benoit Viollet for AMPKα2 KO mice, Dalton Saunders for his help with drug studies and Zu-Xi Yu for assistance with electron microscopy. We also thank Adam Weidenhammer and Yiying Tsai for their technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS: S.J.P. performed majority of experiments and analyzed the data and wrote the paper, O.G. performed hyperinsulinemic-euglycemic clamp study. A.L.B., J.K., X.X, S.Y. and J.H.U. helped with experiments. J.E.S. and E.D.A performed mitochondrial function study. S.B. and R.L.L. performed muscle fiber analysis. L.G.K. and S.L.B. generated the LCR and HCR rats. A.P. and K.B. helped with mitochondria experiments. S.G.K. provided rhesus macaque tissue. M.K.K. and J.H.C. designed and supervised the study, analyzed the data and wrote the manuscript.
References
- Anderson CW, Carter TH. The DNA-activated protein kinase – DNA-PK. Curr Top Microbiol Immunol. 1996;217:91–111. doi: 10.1007/978-3-642-50140-1_7. [DOI] [PubMed] [Google Scholar]
- Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. 2007;132:2131–2157. doi: 10.1053/j.gastro.2007.03.054. [DOI] [PubMed] [Google Scholar]
- Bailey SM, Meyne J, Chen DJ, Kurimasa A, Li GC, Lehnert BE, Goodwin EH. DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proc Natl Acad Sci U S A. 1999;96:14899–14904. doi: 10.1073/pnas.96.26.14899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barazzoni R, Short KR, Nair KS. Effects of aging on mitochondrial DNA copy number and cytochrome c oxidase gene expression in rat skeletal muscle, liver, and heart. J Biol Chem. 2000;275:3343–3347. doi: 10.1074/jbc.275.5.3343. [DOI] [PubMed] [Google Scholar]
- Blunt T, Gell D, Fox M, Taccioli GE, Lehmann AR, Jackson SP, Jeggo PA. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc Natl Acad Sci U S A. 1996;93:10285–10290. doi: 10.1073/pnas.93.19.10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation. 2005;112:2686–2695. doi: 10.1161/CIRCULATIONAHA.105.554360. [DOI] [PubMed] [Google Scholar]
- Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–2466. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- Chen BP, Chan DW, Kobayashi J, Burma S, Asaithamby A, Morotomi-Yano K, Botvinick E, Qin J, Chen DJ. Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem. 2005;280:14709–14715. doi: 10.1074/jbc.M408827200. [DOI] [PubMed] [Google Scholar]
- Critchlow SE, Jackson SP. DNA end-joining: from yeast to man. Trends Biochem Sci. 1998;23:394–398. doi: 10.1016/s0968-0004(98)01284-5. [DOI] [PubMed] [Google Scholar]
- Espejel S, Martin M, Klatt P, Martin-Caballero J, Flores JM, Blasco MA. Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice. EMBO Rep. 2004;5:503–509. doi: 10.1038/sj.embor.7400127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, Sartorelli V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garton FC, North KN, Koch LG, Britton SL, Nogales-Gadea G, Lucia A. Rodent models for resolving extremes of exercise and health. Physiol Genomics. 2016;48:82–92. doi: 10.1152/physiolgenomics.00077.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson RW, Hakimi P. Born to run; the story of the PEPCK-Cmus mouse. Biochimie. 2008;90:838–842. doi: 10.1016/j.biochi.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nature reviews Molecular cell biology. 2007;8:774–785. doi: 10.1038/nrm2249. [DOI] [PubMed] [Google Scholar]
- Harris MI. Undiagnosed NIDDM: clinical and public health issues. Diabetes Care. 1993;16:642–652. doi: 10.2337/diacare.16.4.642. [DOI] [PubMed] [Google Scholar]
- Huston E, Lynch MJ, Mohamed A, Collins DM, Hill EV, MacLeod R, Krause E, Baillie GS, Houslay MD. EPAC and PKA allow cAMP dual control over DNA-PK nuclear translocation. Proc Natl Acad Sci U S A. 2008;105:12791–12796. doi: 10.1073/pnas.0805167105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014;24:464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram DK, Cutler RG, Weindruch R, Renquist DM, Knapka JJ, April M, Belcher CT, Clark MA, Hatcherson CD, Marriott BM, et al. Dietary restriction and aging: the initiation of a primate study. J Gerontol. 1990;45:B148–163. doi: 10.1093/geronj/45.5.b148. [DOI] [PubMed] [Google Scholar]
- Ingram DK, Lane MA, Cutler RG, Roth GS. Longitudinal study of aging in monkeys: effects of diet restriction. Neurobiol Aging. 1993;14:687–688. doi: 10.1016/0197-4580(93)90072-j. [DOI] [PubMed] [Google Scholar]
- Jackson SP. DNA-dependent protein kinase. Int J Biochem Cell Biol. 1997;29:935–938. doi: 10.1016/s1357-2725(97)00006-x. [DOI] [PubMed] [Google Scholar]
- Koch LG, Britton SL, Wisloff U. A rat model system to study complex disease risks, fitness, aging, and longevity. Trends Cardiovasc Med. 2012;22:29–34. doi: 10.1016/j.tcm.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkinos P, Myers J, Kokkinos JP, Pittaras A, Narayan P, Manolis A, Karasik P, Greenberg M, Papademetriou V, Singh S. Exercise capacity and mortality in black and white men. Circulation. 2008;117:614–622. doi: 10.1161/CIRCULATIONAHA.107.734764. [DOI] [PubMed] [Google Scholar]
- Koonen DP, Sung MM, Kao CK, Dolinsky VW, Koves TR, Ilkayeva O, Jacobs RL, Vance DE, Light PE, Muoio DM, et al. Alterations in skeletal muscle fatty acid handling predisposes middle-aged mice to diet-induced insulin resistance. Diabetes. 2010;59:1366–1375. doi: 10.2337/db09-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, et al. Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell metabolism. 2010;12:668–674. doi: 10.1016/j.cmet.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees-Miller SP, Anderson CW. The human double-stranded DNA-activated protein kinase phosphorylates the 90-kDa heat-shock protein, hsp90 alpha at two NH2-terminal threonine residues. J Biol Chem. 1989;264:17275–17280. [PubMed] [Google Scholar]
- Li J, Buchner J. Structure, function and regulation of the hsp90 machinery. Biomed J. 2013;36:106–117. doi: 10.4103/2319-4170.113230. [DOI] [PubMed] [Google Scholar]
- Li M, Lin YF, Palchik GA, Matsunaga S, Wang D, Chen BP. The catalytic subunit of DNA-dependent protein kinase is required for cellular resistance to oxidative stress independent of DNA double-strand break repair. Free Radic Biol Med. 2014;76:278–285. doi: 10.1016/j.freeradbiomed.2014.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O, Michael LF, Puigserver P, Isotani E, Olson EN, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- Liu X, Huh JY, Gong H, Chamberland JP, Brinkoetter MT, Hamnvik OP, Mantzoros CS. Lack of mature lymphocytes results in obese but metabolically healthy mice when fed a high-fat diet. Int J Obes (Lond) 2015;39:1548–1557. doi: 10.1038/ijo.2015.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Lluch G, Hunt N, Jones B, Zhu M, Jamieson H, Hilmer S, Cascajo MV, Allard J, Ingram DK, Navas P, et al. Calorie restriction induces mitochondrial biogenesis and bioenergetic efficiency. Proc Natl Acad Sci U S A. 2006;103:1768–1773. doi: 10.1073/pnas.0510452103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod JN, Shapiro BH. Repetitive blood sampling in unrestrained and unstressed mice using a chronic indwelling right atrial catheterization apparatus. Lab Anim Sci. 1988;38:603–608. [PubMed] [Google Scholar]
- Mason SD, Howlett RA, Kim MJ, Olfert IM, Hogan MC, McNulty W, Hickey RP, Wagner PD, Kahn CR, Giordano FJ, et al. Loss of skeletal muscle HIF-1alpha results in altered exercise endurance. PLoS Biol. 2004;2:e288. doi: 10.1371/journal.pbio.0020288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattison JA, Lane MA, Roth GS, Ingram DK. Calorie restriction in rhesus monkeys. Experimental gerontology. 2003;38:35–46. doi: 10.1016/s0531-5565(02)00146-8. [DOI] [PubMed] [Google Scholar]
- Mishra A, Brown AL, Yao X, Yang S, Park SJ, Liu C, Dagur PK, McCoy JP, Keeran KJ, Nugent GZ, et al. Dendritic cells induce Th2-mediated airway inflammatory responses to house dust mite via DNA-dependent protein kinase. Nat Commun. 2015;6:6224. doi: 10.1038/ncomms7224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405–415. doi: 10.1016/j.cell.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nony P, Gaude H, Rossel M, Fournier L, Rouault JP, Billaud M. Stability of the Peutz-Jeghers syndrome kinase LKB1 requires its binding to the molecular chaperones Hsp90/Cdc37. Oncogene. 2003;22:9165–9175. doi: 10.1038/sj.onc.1207179. [DOI] [PubMed] [Google Scholar]
- O’Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jorgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A. 2011;108:16092–16097. doi: 10.1073/pnas.1105062108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oettinger MA, Schatz DG, Gorka C, Baltimore D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 1990;248:1517–1523. doi: 10.1126/science.2360047. [DOI] [PubMed] [Google Scholar]
- Omodei D, Fontana L. Calorie restriction and prevention of age-associated chronic disease. FEBS Lett. 2011;585:1537–1542. doi: 10.1016/j.febslet.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140–1142. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Pullen L, Bolon DN. Enforced N-domain proximity stimulates Hsp90 ATPase activity and is compatible with function in vivo. The Journal of biological chemistry. 2011;286:11091–11098. doi: 10.1074/jbc.M111.223131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Weiqiang K, Qing Z, Pengju Z, Yi L. Aging impairs insulin-stimulated glucose uptake in rat skeletal muscle via suppressing AMPKalpha. Exp Mol Med. 2007;39:535–543. doi: 10.1038/emm.2007.59. [DOI] [PubMed] [Google Scholar]
- Quanz M, Herbette A, Sayarath M, de Koning L, Dubois T, Sun JS, Dutreix M. Heat shock protein 90alpha (Hsp90alpha) is phosphorylated in response to DNA damage and accumulates in repair foci. The Journal of biological chemistry. 2012;287:8803–8815. doi: 10.1074/jbc.M111.320887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queitsch C, Sangster TA, Lindquist S. Hsp90 as a capacitor of phenotypic variation. Nature. 2002;417:618–624. doi: 10.1038/nature749. [DOI] [PubMed] [Google Scholar]
- Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol. 2009;16:819–824. doi: 10.1038/nsmb.1641. [DOI] [PubMed] [Google Scholar]
- Reznick RM, Zong H, Li J, Morino K, Moore IK, Yu HJ, Liu ZX, Dong J, Mustard KJ, Hawley SA, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell metabolism. 2007;5:151–156. doi: 10.1016/j.cmet.2007.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123:2764–2772. doi: 10.1172/JCI67227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solier S, Kohn KW, Scroggins B, Xu W, Trepel J, Neckers L, Pommier Y. Feature Article: Heat shock protein 90alpha (HSP90alpha), a substrate and chaperone of DNA-PK necessary for the apoptotic response. Proceedings of the National Academy of Sciences of the United States of America. 2012 doi: 10.1073/pnas.1203617109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwa M, Nakano H, Kumagai S. Effects of chronic AICAR treatment on fiber composition, enzyme activity, UCP3, and PGC-1 in rat muscles. J Appl Physiol. 2003;95:960–968. doi: 10.1152/japplphysiol.00349.2003. [DOI] [PubMed] [Google Scholar]
- Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell. 2012;150:987–1001. doi: 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima Y, Gavrilova O, Yakar S, Jou W, Pack S, Asghar Z, Wheeler MB, LeRoith D. Leptin improves insulin resistance and hyperglycemia in a mouse model of type 2 diabetes. Endocrinology. 2005;146:4024–4035. doi: 10.1210/en.2005-0087. [DOI] [PubMed] [Google Scholar]
- van Bergen en Henegouwen PM, Berbers G, Linnemans WA, van Wijk R. Subcellular localization of the 84,000 dalton heat-shock protein in mouse neuroblastoma cells: evidence for a cytoplasmic and nuclear location. Eur J Cell Biol. 1987;43:469–478. [PubMed] [Google Scholar]
- Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RH, Chang I, Hudak CS, Hyun S, Kwan HY, Sul HS. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell. 2009;136:1056–1072. doi: 10.1016/j.cell.2008.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn JH, Kim JK, Buchanan TA. Time courses of changes in hepatic and skeletal muscle insulin action and GLUT4 protein in skeletal muscle after STZ injection. Diabetes. 1994;43:564–571. doi: 10.2337/diab.43.4.564. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.