

Abstract
Objectives
To determine the incidence of pathogenic SCN8A variants in a cohort of epilepsy patients referred for clinical genetic testing. We also investigated the contribution of SCN8A to autism spectrum disorder, intellectual disability, and neuromuscular disorders in individuals referred for clinical genetic testing at the same testing laboratory.
Methods
Sequence data from 275 epilepsy panels screened by Emory Genetics Laboratory were reviewed for variants in SCN8A. Additional cases with variants in SCN8A were ascertained from other testing laboratories. Parental samples were tested for variant segregation and clinical histories were examined. SCN8A variants detected from gene panel analyses for autism spectrum disorder, intellectual disability, and neuromuscular disorders were also examined.
Results
Five variants in SCN8A were identified in five individuals with epilepsy. Three variants were de novo, one was inherited from an affected parent, and one was inherited from an unaffected parent. Four of the individuals have epilepsy and developmental delay/intellectual disability. The remaining individual has a milder epilepsy presentation without cognitive impairment. We also identified an amino acid substitution at an evolutionarily conserved SCN8A residue in a patient who was screened on the autism spectrum disorder panel. Additionally, we examined the distribution of pathogenic SCN8A variants across the Nav1.6 channel and identified four distinct clusters of variants. These clusters are primarily located in regions of the channel that are important for the kinetics of channel inactivation.
Conclusions
Variants in SCN8A may be responsible for a spectrum of epilepsies as well as other neurodevelopmental disorders without seizures. The predominant pathogenic mechanism appears to involve disruption of channel inactivation, leading to gain-of-function effects.
Keywords: SCN8A, Sodium channels, Epilepsy, Gene panel analysis
1. Introduction
Voltage-gated sodium channels (VGSCs) are important regulators of neuronal excitability. As a result, pathogenic variants in VGSCs are responsible for a number of pathophysiological conditions, particularly epilepsy. The VGSC SCN8A encodes the pore-forming α-subunit Nav1.6, which is expressed in excitatory and inhibitory neurons throughout the central and peripheral nervous systems (Caldwell et al., 2000; Oliva et al., 2012).
VGSC α-subunits are made up of four homologous domains (DI-DIV), each containing six transmembrane segments (S1–S6). Mutations in the mouse homolog of SCN8A (Scn8a) were originally associated with motor disorders based on the phenotypes of more than ten different mouse lines with null or hypomorphic alleles of Scn8a. Scn8a mouse mutants exhibit a range of phenotypes, including ataxia, tremor, dystonia, hind limb paralysis, and premature lethality with recessive inheritance (Meisler et al., 2001; O’Brien and Meisler, 2013). These mice do not exhibit spontaneous convulsive seizures, and furthermore, mice heterozygous for these mutant alleles were found to be more resistant to chemically and electrically induced seizures (Makinson et al., 2014; Martin et al., 2007). In 2006 the identification of a heterozygous, frameshift SCN8A variant in a patient exhibiting intellectual disability, cerebellar atrophy, and ataxia was consistent with the reported mouse models of Scn8a dysfunction (Trudeau et al., 2006).
The first SCN8A epilepsy mutation was identified in 2012 by whole-genome sequencing (WGS) in a patient with severe epileptic encephalopathy who exhibited early-onset seizures, autistic features, intellectual disability, ataxia, and sudden unexpected death in epilepsy (SUDEP) (Veeramah et al., 2012). The heterozygous missense variant, p.N1768D, was determined to be de novo in the patient. Since this initial discovery, there has been a sharp rise in the number of identified pathogenic SCN8A variants in patients with epilepsy, with over one hundred mutations reported to date (Meisler et al., 2016). Most of the SCN8A variants have been detected in individuals with early infantile epileptic encephalopathy (EIEE), similar to the initial patient. Furthermore, nearly all reported mutations are missense variants that were de novo or inherited from an unaffected parent who was found to be mosaic. Functional analysis of eight variants revealed gain-of-function effects as the predominant pathogenic mechanism, although two of the variants produced apparent loss-of-function effects in vitro (Blanchard et al., 2015; de Kovel et al., 2014; Estacion et al., 2014; Veeramah et al., 2012; Wagnon et al., 2016). In 2015, Wagnon et al. reported the generation of a mouse expressing the p.N1768D mutation in the orthologous Scn8a gene. Unlike previous mouse lines, these mutants exhibit spontaneous seizures and premature lethality (Wagnon et al., 2015).
Recently, Gardella et al. reported three unrelated families carrying the same SCN8A missense variant, p.E1483K, in the inactivation gate of the channel (Gardella et al., 2016). Interestingly, all three families exhibited benign infantile seizures. This was the first report of SCN8A-associated epilepsy that showed a more benign seizure course without intellectual disability. The authors speculated that this variant in the inactivation gate might have a more modest effect on channel inactivation, thereby resulting in the milder phenotype seen in the three families.
While the application of whole-genome and -exome sequencing to severe epilepsy cases was essential to the discovery of the initial SCN8A pathogenic variants, many variants are now being identified via gene panel analysis (also known as targeted resequencing). One advantage of targeted gene panel analysis over whole-exome sequencing is that read coverage is increased across the genes of interest, thus reducing the possibility that clinically important variants in these genes are missed. More than 15 commercial epilepsy panels that include sequencing of SCN8A are currently available.
Here we report five new SCN8A epilepsy variants identified via gene panel analysis. We report several cases that have phenotypes consistent with previous reports for SCN8A-encephalopathy, as well as one case with an inherited SCN8A variant and a milder epilepsy presentation.
2. Material and methods
2.1. Patients
Sequence data from 275 patients screened by the Emory Genetics Laboratory (EGL) using the Epilepsy and Seizure Disorders (ESD) panel were reviewed for this study. Two additional patients with SCN8A variants had targeted gene panel testing performed by other diagnostic laboratories (GeneDx and Athena Diagnostics). All patients had epilepsy as one of the indications for genetic testing. Peripheral blood samples were obtained from family members to test variant inheritance after written consent was obtained. Clinical information for each patient was obtained from their corresponding clinicians. This study was approved by the Institutional Review Board of Emory University.
2.2. Targeted gene panel sequencing
At EGL, a custom-designed in-solution hybridization probe library (IDT or SureSelect, Agilent Technologies, Santa Clara, CA) was used to capture the coding exons of 110 genes, including SCN8A, on the Epilepsy and Seizure Disorders (ESD) panel. Direct sequencing of the amplified captured regions was performed using next-generation sequencing (2x100bp, paired end reads) on an Illumina HiSeq 2500 (Illumina, San Diego, CA) in rapid run mode. The individual DNA sequence reads were aligned to the published human genome reference (hg19 build) and variants were called using NextGENe® (SoftGenetics, State College, PA). Further analysis was performed using the EGL bioinformatics pipeline which annotates the identified variants utilizing a variety of external and internal sources. Variants are called within the coding exons and +/− 10bp into the introns. Relevant regions of epilepsy genes not amenable to NGS were filled in using the Sanger sequencing method.
2.3. Sanger sequencing
All SCN8A variants were confirmed by Sanger sequencing. Parent and sibling samples were also Sanger sequenced to determine whether the SCN8A variant segregated with disease or arose de novo.
2.4. Deep amplicon sequencing
Sequencing of a 392-base pair amplicon encompassing the SCN8A c.2287A>G (p.I763V) variant from Patient 1 and his parents to approximately 400,000X coverage was performed by the Emory Integrated Genomics Core (EIGC).
3. Results
3.1. Identification of novel SCN8A epilepsy mutations
All patients had seizure onset within the first year of life and were referred for genetic testing. The identified SCN8A amino acid substitutions were distributed across the channel: one in the N-terminal domain (p.K101R), one in the DIIS1 (p.I763V), one in the linker between DIIIS4 and DIIIS5 (p.N1329D), one in DIIIS5 (p.L1332R), and one in the C-terminal domain (p.N1877S) (Figure 1A). All five mutations occur at evolutionarily conserved amino acid positions (Figure 1B) and were predicted to be damaging to protein function by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/) prediction algorithms. Table 1 summarizes the clinical features of the patients.
Figure 1. Location within the Nav1.6 channel (A) and evolutionary conservation (B) of the SCN8A variants examined in this study.

(A) Location of the variants reported in this study in the Nav1.6 channel. Roman numerals correspond to the four repeat domains of the channel (DI–DIV). Cylinders represent the six transmembrane segments of each domain. Filled circles denote de novo epilepsy variants. Open circles indicate inherited epilepsy variants. The grey triangle indicates a variant detected from an autism panel. (B) Protein alignment of the human, mouse, rat, zebrafish, and fruit fly VGSC α-subunits. hNav1.6: human, mNav1.6: mouse, rNav1.6: rat, zNav1.6: zebrafish, Dm para: Drosophila melanogaster.
Table 1.
Clinical characteristics of epilepsy patients with SCN8A variants
| Case | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 |
|---|---|---|---|---|---|
| Sex | Male | Female | Male | Male | Female |
| Age | 5 years | 4 years | 11 months | 6 years | 15 years |
| Nucleotide change | c.2287A>G | c.3985A>G | c.3995T>G | c.5630A>G | c.302A>G |
| Protein change | p.I763V | p.N1329D | p.L1332R | p.N1877S | p.K101R |
| Inheritance | de novo | de novo | de novo | Inherited from affected father | Inherited from unaffected mother |
| Variant Classification | Pathogenic | Pathogenic | Pathogenic | Likely Pathogenic | Variant of Unknown Significance |
| Family history | Affected fraternal twin brother | none | none | Affected brother and father | none |
| Age at onset | 4 months | 3 months | 1 month | 9 months | 1 month |
| Diagnosis | Intractable epilepsy | Epileptic encephalopathy | Infantile spasms | Nocturnal Partial Complex seizures with secondary generalization | Seizures |
| Development | Delayed, especially speech and language | Global developmental delay | Developmental delay | Normal | Developmental delay, intellectual disability, speech delay |
| EEG | Mild generalized slowing of background with rare left frontal epileptiform discharges | Diffuse cerebral dysfunction, slowing and disorganization of background, multifocal epileptiform discharges | Background disorganization and slowing, multiform epileptiform discharges | Near continuous sleep enhanced spike and wave activity, generalized or shifting predominance between left and right anterior areas | Sharp spikes and waves in sleep |
| MRI | Linear areas of decreased signal from chronic subarachnoid hemorrhage | Mild cerebral volume loss | Left frontal lobe cortical dysplasia | Normal | Inferior vermis hypoplasia |
| Motor development | Mildly delayed | Wheelchair bound, minimal head control | Delayed | Normal | --- |
| Other phenotypes | Obstructive sleep apnea, subdural hematoma, status epilepticus | Hypotonia, poor vision, nystagmus, feeding difficulties, GERD | Hypotonia, GERD, brachycephaly, nystagmus | --- | Hypotonia, precocious puberty, short stature, facial dysmorphism |
| Response to treatment | Refractory | Refractory | Refractory | Seizure-free for 3 years on lacosamide but seizures reoccurred. No response to levetiracetam, valproic acid. Marked improvement on phenytoin | Seizure-free for several years on oxcarbazepine but continues medication |
Variants are annotated according to the reference transcript NM_014191.3.
3.2. Patient Descriptions
Patient 1: SCN8A, c.2287A>G, p.I763V
Patient 1 presented with seizures at four months of age and was recognized to have a subarachnoid hemorrhage. The hemorrhage was drained via a burr hole; however, he continued to have multiple generalized tonic-clonic seizures requiring over 20 hospitalizations, and at least one instance of status epilepticus. He is developmentally delayed, especially for speech and language. Electroencephalograms (EEGs) show a mild generalized slowing of the background with rare left frontal epileptiform discharges. Magnetic resonance imaging (MRI) showed areas of decreased linear signal as a result of the subarachnoid hemorrhage. Interestingly, the individual has a fraternal twin brother who had a cystic subdural hygroma and is similarly affected by seizures and developmental delays.
Genetic testing of Patient 1 at EGL uncovered heterozygous variants in four known epilepsy genes: PRICKLE1 (c.2002T>A, p.S668T), KCNQ3 (c.1720C>T, p.P574S), and SCN8A (c.2287A>G, p.I763V), as well as a deletion at 2q23.1 encompassing noncoding exon 5 of the MBD5 gene (chr2: 149,057,023-149,166,288) (hg19). The PRICKLE1 variant was not tested further since PRICKLE1 is associated with autosomal recessive disease, and only one variant was detected in the patient. Parental testing detected the MBD5 deletion in the unaffected mother and the KCNQ3 variant in the unaffected father. The KCNQ3 variant has also been observed 242 times in the ExAC database, indicating that it is likely to be a benign variant. In contrast, neither parent carried the SCN8A variant. The affected twin brother was also found to carry the MBD5 deletion and the SCN8A missense variant. SCN8A p.I763V is located within the DIIS1 transmembrane domain near the previously published T767I epilepsy mutation (Estacion et al., 2014). The isoleucine residue is evolutionarily invariant in all members of the VGSC family (Fig. 1), and the change to valine is predicted to be damaging by PolyPhen-2 and SIFT.
Because the SCN8A variant was detected in both the patient and his brother but was absent in the parents, we attempted to determine whether one of the parents was mosaic. However, we could not detect the variant in DNA extracted from blood by deep amplicon sequencing in either parent (up to 400,000X coverage, data not shown). We conclude that mosaicism is likely to be limited to the germline of one of the parents. This variant has not been observed in the ExAC database but has been reported once as a variant of unknown significance (VUS) from an epilepsy patient by GeneDx in ClinVar. Although we believe that the SCN8A variant is the causative mutation in these two brothers, we cannot rule out the possibility that the deletion of MBD5 noncoding exon 5 may also contribute to the disease presentation in these individuals since heterozygous deletions of MBD5 are associated with developmental delay, seizures, and language impairment due to haploinsufficiency. While deletions confined to the noncoding exons of MBD5 (exons 1–5) have been reported in disease previously (Talkowski et al., 2011), none have been limited to exon 5 as in Patient 1, making it difficult to compare across cases. Exon 5 is the most proximal noncoding exon to the first coding exon (exon 6), however, this region is not evolutionary conserved and the deletion was inherited from an unaffected parent.
Patient 2: SCN8A, c.3985A>G, p.N1329D
Patient 2 exhibited normal development until seizure onset at three months of age. She subsequently developed refractory epilepsy, global developmental delays, poor vision and nystagmus, and feeding difficulties and gastroesophageal reflux disease (GERD). EEG showed diffuse cerebral dysfunction and slowing and disorganization of background with multifocal epileptiform discharges. MRI showed mild loss of cerebral volume. Previous genetic testing included a chromosomal microarray, an Infantile Epilepsy Panel, fragile X FMR1 testing, and sequencing of NPC1 and NPC2. NPC2 sequencing revealed a heterozygous variant (c.352G>A, p.E118K), and the Infantile Epilepsy Panel uncovered a heterozygous variant of uncertain significance in ALDH7A1 (c.235A>G, p.R79G). Since neither variant was thought to contribute to the patient’s clinical phenotype, the ESD panel was ordered at EGL. This test not only confirmed the presence of the ALDH7A1 variant but also identified a heterozygous variant in SCN8A (c.3985A>G, p.N1329D) and a heterozygous intronic duplication in CNTNAP2. Parental testing revealed that the SCN8A variant was de novo in the patient, and parental identity was confirmed with microsatellite analysis. ALDH7A1 and CNTNAP2 were excluded from further analysis since these genes are associated with autosomal recessive disorders and were not consistent with the phenotype. SCN8A p.N1329D has not been seen before in disease or in the ExAC database. Furthermore, it is located in the intracellular linker between DIIIS4 and DIIIS5, in which several pathogenic variants have already been identified, including p.I1327V and p.L1331V. N1329 is evolutionarily invariant across species and the other VGSCs, and the change to a negatively charged aspartic acid is predicted to be damaging.
Patient 3: SCN8A, c.3995T>G, p.L1332R
Patient 3 presented with abnormal involuntary movements at one month of age, and EEG analysis revealed hypsarrhythmia suggestive of infantile spasms. EEG at two months showed mild background slowing and disorganization with multifocal epileptiform discharges consistent with epileptic encephalopathy. He displays global developmental delays, hypotonia, nystagmus, and brachycephaly. MRI showed left frontal lobe cortical dysplasia. Chromosomal microarray analysis was normal. Epilepsy gene panel testing performed by GeneDx (Infantile Epilepsy panel) identified heterozygous variants in SCN8A (c.3995T>G, p.L1332R) and KCNJ10 (c.1043G>A, p.R348H). Testing of the parents did not uncover the SCN8A variant, indicating that the variant arose de novo in the patient. The KCNJ10 variant was not tested since it is associated with autosomal recessive disease and only one variant was detected. SCN8A p.L1332R has not been seen previously in disease or in the ExAC database, but is directly adjacent to the reported pathogenic variant, p.L1331V, in the DIIIS5 transmembrane domain of the channel. The p.L1332R substitution affects an evolutionarily invariant residue and is predicted to be damaging by PolyPhen-2 and SIFT.
Patient 4: SCN8A, c.5630A>G, p.N1877S
Patient 4 presented with seizures at nine months of age, which included staring spells during wakefulness and later included generalized tonic-clonic seizures during sleep. He was started on oxcarbazepine, which controlled his father’s seizures, but developed a severe allergic reaction. Levetiracetam was ineffective. Seizures were fully controlled for three years on lacosamide but then recurred. He continued to have 1–2 seizures during sleep per month despite treatment. Initial EEG was normal. EEG at age six years revealed very active, sleep enhanced spike and wave activity, which was generalized or shifting predominance between left and right anterior areas. The individual performs reasonably well in school but is somewhat behind his twin sister. He has had marked improvement, but not complete control, on phenytoin.
The individual has a family history of epilepsy. His father (Figure 2, I:1) started having seizures at six months of age and continued to have frequent seizures despite treatment with phenobarbital until age six, at which point his seizures improved. At age 13, treatment was switched to carbamazepine, from which point he continued to have seizures about twice a year. Seizures stopped in his late teens, but he has had seizures on several occasions when he tried to stop taking his medication. All EEGs and MRIs were reported to be normal, and the father has normal cognition. Patient 4 also has a younger brother (Figure 2, II:3) who presented with seizures at 7 months of age. The younger brother’s EEG was normal and his seizures are fully controlled on oxcarbazepine. An epilepsy gene panel performed by Athena Diagnostics (Epilepsy Advanced Sequencing Evaluation) identified a number of heterozygous single nucleotide variants in the younger brother, including SCN8A (c.5630A>G, p.N1877S), GRIN2A (c.4353A>T, p.R1451S), TSC1 (c.1631G>A, p.G544E), ATP2A2 (c.1912A>G, p.I638V), and LBR (c.899A>G, p.Y300C). We performed familial segregation analysis for the SCN8A, GRIN2A, ATP2A2, and TSC1 variants (LBR was excluded since seizures have only been seen in homozygous patients and only one variant was identified from panel testing). The SCN8A variant was present in all three affected family members, but was absent from the unaffected mother (I:2) and the patient’s unaffected twin (II:1). The GRIN2A variant was present in the father (I:1) and younger brother (II:3) but absent from Patient 4 (II:2). The ATP2A2 variant was present in the unaffected mother (I:2), unaffected sister (II:1), and the younger brother (II:3). Finally, the TSC1 variant was detected in the mother and both her sons. This pattern of inheritance lends support to the SCN8A variant being causative, although we cannot rule out the possibility that the GRIN2A variant carried by the father and the younger brother may act as a modifier, decreasing the severity of their epilepsy and the EEG abnormalities.
Figure 2. Pedigree of Patient 4 showing the inheritance pattern of the SCN8A and GRIN2A variants.
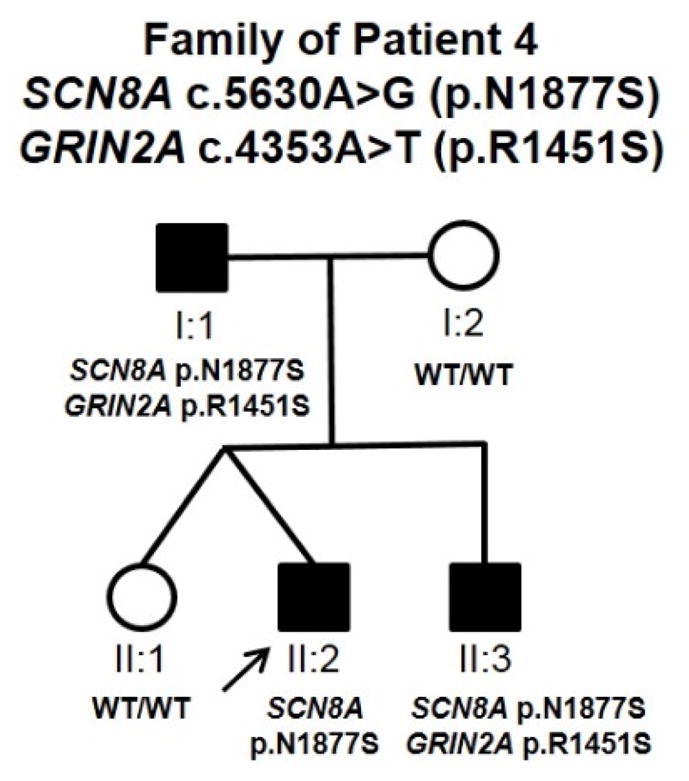
Filled symbols indicate affected individuals. The arrow denotes Patient 4.
The SCN8A p.N1877S variant resides in the C-terminal domain of the channel, proximal to the mutational hotspot p.R1872, at which three different amino acid substitutions have been identified in ten unrelated epilepsy patients (Wagnon et al., 2016). N1877S is predicted to be damaging by PolyPhen-2 and SIFT. SCN8A p.N1877S has not been observed in the ExAC database but has been reported twice in ClinVar (once as a VUS and once as a likely pathogenic de novo variant) and has recently been reported in a father and son with early-onset epilepsy without cognitive impairment (Anand et al., 2016). This additional instance of parental transmission and milder epilepsy presentation associated with the same SCN8A variant again lends support to the prediction that this is a pathogenic variant.
Patient 5: SCN8A, c.302A>G, p.K101R
Seizure onset in Patient 5 began in the neonatal period, and seizures occurred approximately once a year between ages one and four. At age seven, she began to have more frequent seizures, which were controlled on oxcarbazepine. Staring spells were also reported. The individual’s history is significant for developmental delay, aphasia, hypotonia, short stature, and moderate to severe cognitive impairment. Patient 5 also exhibits craniofacial abnormalities, including brachycephaly with a broad, tall forehead, bilateral epicanthal folds, flattening of the nasal bridge, low hairline, very full lips, and a high arched palate. EEG revealed sharp waves and spikes in sleep; however, this activity was brief and did not correlate with any clinical events. MRI showed the stable appearance of an inferior vermian hypoplasia. Prior genetic testing included two microarrays to detect unbalanced chromosomal abnormalities, UBE3A sequence and deletion/duplication analysis, and SNRPN methylation analysis, all of which were normal. Testing on EGL’s ESD panel uncovered a heterozygous variant in the N-terminal domain of SCN8A (c.302A>G, p.K101R). K101 is evolutionarily invariant, and the identified substitution is predicted to be damaging by PolyPhen-2 and SIFT. Parental testing revealed that the variant was inherited from her unaffected mother. The p.K101R variant was also reported once in the ExAC database. The presence of this variant in an unaffected parent raises two possibilities: the variant might not be the cause of disease in Patient 5, or this variant may exhibit reduced penetrance in the mother.
Further sequence analysis revealed the presence of a known pathogenic variant in the PACS1 gene (c.607C>T, p.R203W) in Patient 5. PACS1 p.R203W has been reported previously as a de novo variant from whole-exome sequencing in three unrelated male patients with intellectual disability and similar facial dysmorphisms (Gadzicki et al., 2015; Helbig et al., 2016). Two of these patients had seizures in the neonatal period similar to Patient 5. Parental testing did not detect the PACS1 variant in the mother; the father was unavailable for testing. PACS1 p.R203W has also been reported once in the ExAC database.
3.3. SCN8A variation in other neurodevelopmental disorders
In addition to seizures, individuals with SCN8A encephalopathy often experience other neurodevelopmental abnormalities, including intellectual disability, autism, and movement disorders such as ataxia and dystonia (Larsen et al., 2015). To investigate whether variation in SCN8A contributes to these other neurodevelopmental disorders with or without seizures, we examined variants compiled from gene panel testing at EGL for autism spectrum disorder, intellectual disability, and neuromuscular disorders. A total of 107 SCN8A variants were detected from 91 patients (52 autism referrals, 26 intellectual disability referrals, and 13 neuromuscular referrals). Seventy-two variants (67%) were located in intronic sequences, none of which affected consensus splice sites. Twenty-three variants (22%) were synonymous substitutions and were not predicted to affect the protein sequence or splicing. Twelve patients (13%) were found to have missense substitutions that altered the amino acid sequence of the protein. The polymorphisms c.3076C>T (p.R1026C) and c.2098A>T (p.I700L) were identified in six individuals and one individual, respectively (Table 2). Of the remaining five missense variants, only one (p.I805M) altered an evolutionarily conserved residue and was predicted to be damaging by in silico analysis (Adzhubei et al., 2010). I805M is located within the DIIS2 transmembrane domain, and substitution of this amino acid has not been observed in ExAC or in any other database (Figure 1). This variant was identified from an autism spectrum disorder referral; however, we do not have information on the clinical presentation of the patient. Three of the other four rare missense variants (p.I68V, p.G1050S, p.R1960Q) have been reported before in the ClinVar database. The N-terminal variant (p.I68V) was reported as a likely benign variant by the Genetics Services Laboratory at University of Chicago. The C-terminal variant (p.R1960Q) was reported as a VUS from an infant-epilepsy panel by GeneDx and has been seen three times in the ExAC database. The p.G1050S variant was reported previously as a VUS from an epilepsy panel by GeneDx, once as a likely pathogenic de novo variant in a patient with hemiplegic cerebral palsy and intellectual disability, and ten times in the ExAC database (McMichael et al., 2015). At this time, it is unclear whether these rare variants contribute to disease or represent benign variants. SCN8A may therefore contribute to other neurodevelopmental disorders, and additional screening for SCN8A mutations should be conducted to explore the full spectrum of phenotypes associated with SCN8A dysfunction.
Table 2.
SCN8A variants identified from panel testing for other neurodevelopmental disorders
| Gene | Variant | Protein | No. Obs. | Location in Channel | Evolutionarily Conserved | Controls (No.) | Gene Panel |
|---|---|---|---|---|---|---|---|
| SCN8A | c.202A>G | p.I68V | 1 | N-terminus | No | No | Neuromuscular panel |
| SCN8A | c.2098A>T | p.I700L | 1 | Cytoplasmic Loop 1 | No | ExAC (315) | ID panel |
| SCN8A | c.2415A>G | p.I805M | 1 | DIIS2 | Yes | No | ASD panel |
| SCN8A | c.3076C>T | p.R1026C | 6 | Cytoplasmic Loop 2 | No | ExAC (1418) | ASD and ID panels |
| SCN8A | c.3097C>G | p.P1033A | 1 | Cytoplasmic Loop 2 | No | No | ASD panel |
| SCN8A | c.3148G>A | p.G1050S | 1 | Cytoplasmic Loop 2 | No | ExAC (10) | ASD panel |
| SCN8A | c.5879G>A | p.R1960Q | 1 | C-terminus | No | ExAC (3) | ASD panel |
Abbreviations: ASD, Autism spectrum disorder; ID, intellectual disability.
Variants are annotated according to the reference transcript NM_014191.3.
3.4. Mutation clusters within the Nav1.6 channel
When the locations of all the reported SCN8A epilepsy variants are overlaid on the Nav1.6 channel, several patterns emerge (Figure 3). Similar to reports for the other VGSCs, many variants reside within the transmembrane domains of the channel, especially the voltage-sensing S4 segment of each repeat domain; however, several intriguing clusters of variants are observed outside of the transmembrane domains. One of these clusters involves the extracellular linker between DIS3 and DIS4 (Figure 3, cluster A). Interestingly, this region of the channel is encoded by two mutually exclusive, alternatively spliced exon 5s (5N and 5A). The usage of these exons is developmentally regulated, with preferential usage of exon 5N early in development and exon 5A during adulthood. Inclusion of each exon has been reported to differentially alter the excitability of the encoded channel (Fletcher et al., 2011; Gazina et al., 2010; Raymond et al., 2004). To date, all the pathogenic variants identified in this region are located in exon 5N. Another observed cluster of variants is located in the 19 amino acid linker between the DIIIS4 and DIIIS5 (cluster B). Clustering of pathogenic variants in this region has also been observed for the Nav1.2 channel (Howell et al., 2015). This linker region is reported to participate in channel inactivation through stabilizing interactions with the nearby inactivation gate (Smith and Goldin, 1997). Consistent with this, we also see a cluster of pathogenic SCN8A variants within the inactivation gate, which is located between DIIIS6 and DIVS1 (cluster C). Finally, there appears to be a cluster of pathogenic variants in the C-terminus, including the recurrently mutated position at p.R1872 (cluster D). Previous electrophysiological analysis of substitutions at R1872 revealed a delay in the inactivation of the mutant channels, which is predicted to increase channel activity (Wagnon et al., 2016).
Figure 3. Distribution of pathogenic epilepsy variants across the Nav1.6 α-subunit.

The filled circles denote the location of pathogenic epilepsy variants reported to be de novo. The open circles denote inherited variants. The four clusters are indicated by lettered circles with arrows.
4. Discussion
The list of SCN8A epilepsy variants has been rapidly growing since the first report in 2012, due in part to the wider availability of genetic testing that includes the SCN8A gene. Pathogenic SCN8A variants appear to account for approximately 1% of patients with epileptic encephalopathies, with over a hundred pathogenic variants identified in SCN8A to date (Meisler et al., 2016). Similar to previous observations, we estimate that variants in SCN8A are responsible for disease in approximately 1% of the cohort of 275 epilepsy patients screened by the Emory Genetics Laboratory, although this cohort is not limited to the epileptic encephalopathies. Pathogenic SCN8A variants are predominantly de novo or inherited from an unaffected parent found to be mosaic. This was true for three of the variants reported in this study (Patients 1–3). Although most pathogenic SCN8A variants are found in individuals with early infantile epileptic encephalopathy (EIEE), an inherited missense variant (p.E1483K) was recently identified in three unrelated families with benign infantile spasms and paroxysmal dyskinesia (Gardella et al., 2016).
In a recent report, Anand and colleagues described a heterozygous SCN8A variant, p.N1877S, in an affected father and son that experienced seizure onset at four and five months of age, respectively, and presented with focal and generalized tonic-clonic seizures. However, neither individual exhibited cognitive impairment. The son’s EEG showed background slowing and disorganization with active focal epileptiform discharges, but his seizures were controlled on carbamazepine (Anand et al., 2016). Here we report an additional family segregating the SCN8A p.N1877S variant. Similar to the family presented by Anand et al, the family described here appears to have a milder epilepsy presentation without cognitive impairment and shows response to treatment with sodium channel blockers such as oxcarbazepine and phenytoin. In contrast to these two families, SCN8A p.N1877S has also been observed de novo in at least two other epilepsy patients with developmental delay and intellectual disability. Anand and colleagues speculated that additional modifying genetic variants may explain the reduced disease severity in their two cases, although no candidates were revealed by the gene panel analysis (Anand et al., 2016). In the family presented here, variants in additional genes were uncovered from the gene panel analysis. This included the NMDA receptor gene, GRIN2A, which is associated with a broad spectrum of epilepsy and speech disorders (Carvill et al., 2013). The C-terminal GRIN2A p.R1451S variant is predicted to be tolerated by in silico analysis and was observed 12 times in the ExAC database. We found that the two family members with p.R1451S (Figure 2, I:1 and II:3) have milder epilepsy presentations and normal EEGs compared to the proband with only the SCN8A variant (II:2). Further studies are needed to determine the functional effects of the SCN8A p.N1877S variant, as well as whether this GRIN2A variant is capable of modifying those effects.
Our last case (Patient 5) highlights the challenges associated with variant interpretation from gene panel analysis. The SCN8A p.K101R variant was the only reported finding from the 110 genes of the ESD panel analysis. This very rare variant (seen in only 1/120,220 alleles in ExAC) was predicted to be damaging by MutationTaster and SIFT (and possibly damaging by PolyPhen-2), and the phenotype of the patient overlaps with what has been reported previously for SCN8A-assoicated epilepsy. Parental testing can be critical for distinguishing pathogenic variants from rare benign familial variants. In this case, the SCN8A variant was inherited from the unaffected mother, suggesting that p.K101R may represent a rare benign variant. The identification of a second variant predicted to be pathogenic (PACS1 p.R203W) also raises the possibility that the SCN8A p.K101R variant may not be pathogenic in this individual. Functional analysis will be necessary to determine whether the p.K101R variant alters the electrophysiological properties of the Nav1.6 channel.
The VGSC genes SCN1A and SCN2A, encoding the Nav1.1 and 1.2 sodium channels, respectively, are associated with a spectrum of epileptic phenotypes, including severe sporadic EIEE and milder familial epilepsies. Mutations in SCN1A were first identified in families with genetic epilepsy with febrile seizures plus (GEFS+) and later recognized as a cause of the catastrophic EIEE Dravet syndrome (Claes et al., 2001; Escayg et al., 2000). Similarly, mutations in SCN2A were originally associated with benign familial neonatal-infantile seizures (BFNIS) and GEFS+, but were later identified in patients with EIEEs, such as Ohtahara syndrome and Dravet syndrome (Berkovic et al., 2004; Nakamura et al., 2013). Consistent with these observations, SCN8A mutations also appear to underlie a spectrum of epilepsy phenotypes.
A small number of SCN8A variants have also been found in individuals with intellectual disability, ataxia, and cerebral palsy, without seizures (McMichael et al., 2015; Rauch et al., 2012; Trudeau et al., 2006). Here we identified an SCN8A missense variant at an evolutionarily conserved amino acid position from a patient with autism. Although we do not know the inheritance of the variant or whether this individual also experienced seizures, autism spectrum disorder is a common comorbidity in patients with pathogenic SCN8A variants (Larsen et al., 2015). Additionally, pathogenic SCN8A variants were recently detected from diagnostic whole-exome sequencing in patients with neurodevelopmental disorders without seizures (Helbig et al., 2016). This is similar to findings for SCN1A and SCN2A, both of which are implicated in autism with and without seizures (Li et al., 2015; O’Roak et al., 2011; Sanders et al., 2012). Consequently, it may be beneficial to add SCN8A to gene panels designed for intellectual disability, autism, and movement disorders, since these disorders share underlying genetic pathways with epilepsy (Li et al., 2015).
To date, five epilepsy variants have been identified in the region of SCN8A encoded by the two alternate exon 5s (5N and 5A). All five of these amino acid substitutions are located within exon 5N (de Kovel et al., 2014; Epi et al., 2013; Larsen et al., 2015; Mercimek-Mahmutoglu et al., 2015; Ohba et al., 2014). Studies in mice and non-human primates (Macaca fascicularis) have shown that the 5N exon, referred to as the neonatal isoform, is expressed early in development and is gradually replaced by isoforms containing the 5A exon (adult isoform) as development progresses (Gazina et al., 2010; Raymond et al., 2004). Little is currently known about the developmental expression pattern of these two exons in humans, although Raymond et al. reported that the 5N exon was observed at higher levels in fetal brain, whereas the 5A exon was incorporated more frequently in transcripts from adult brain (Raymond et al., 2004). The 5N and 5A exons are both 92 bp in length (encoding 30 amino acids) and are separated by a short 155 bp intron. The exons differ at 19 nucleotide positions but only two amino acid positions (p.207I>V and p.212N>D) (Raymond et al., 2004). The usage of these two alternatively spliced exons, encoding a portion of the first domain, is evolutionarily conserved across most of the VGSC α-subunits, including all of the human VGSCs expressed in the CNS. Several studies have examined the functional differences between the two corresponding alternatively spliced isoforms in Nav1.1 and Nav1.2. Electrophysiological studies revealed that alternative splicing of exon 5 modifies the inactivation kinetics of the sodium channel, such that isoforms including the 5N exon inactivate more rapidly compared to those with the 5A exon (Fletcher et al., 2011; Xu et al., 2007). Additionally, the “neonatal” Nav1.2 isoform appears to reduce neuronal excitability in mice (Gazina et al., 2015). Taken together, we hypothesize that mutations in the 5N exon of SCN8A could cause disease by altering the inactivation kinetics of the Nav1.6 channels expressed in early development, leading to hyperexcitability in neurons expressing those channels.
Thus far, there have been no reports of pathogenic variants in the 5A exon. The lack of pathogenic variants in 5A could reflect differences in the electrophysiological properties of the different channel isoforms, such that substitutions in the adult isoform are less detrimental than substitutions in the neonatal isoform. However, when the two exons are examined in the ExAC database, four missense changes are seen in the 5N exon, and only one missense change for the 5A exon. This high level of conservation would suggest both exons are important for proper channel function. Alternatively, the lack of 5A mutations could be the result of the use of sequencing libraries that do not target both SCN8A exons. The current reference transcript for SCN8A (NM_014191.3), used by most laboratories for mutation identification, includes only the 5N exon. As a result, variants in the 5A exon might be missed. At this time, it is unclear whether variants in the 5A exon of SCN8A can also cause disease and, if so, whether disease severity would be similar to that reported for pathogenic variants in other regions of the channel. Mutations in the 5N exon, which is the predominant isoform during early development, lead to an early-onset epilepsy. It is possible that mutations in the 5A exon, expressed later in development, could cause epilepsy with a later age of onset. Interestingly, pathogenic variants have been observed in both alternate exons of SCN2A (denoted as exons 6N and 6A) (Kodera et al., 2013; Nakamura et al., 2013). In these cases, the disease severity did not appear to correlate with the affected exon.
The other three observed clusters of SCN8A variants affect regions of the sodium channel known to have roles in channel inactivation. Two of these clusters, the inactivation gate and the proximal portion of the C-terminus, were noted previously as being enriched for pathogenic variants (Wagnon and Meisler, 2015). The third cluster in the DIIIS4–S5 linker region (Figure 3, cluster B) has not been reported before. This linker is approximately 19 amino acids in length, and five of these positions are reported to be mutated in epilepsy. Two additional epilepsy variants are found directly adjacent to this linker in the DIIIS5 transmembrane domain. This highly conserved linker is known to interact with the inactivation gate by serving as a docking site to stabilize the gating particle in VGSCs. Specifically, the alanine residue at position 1319 of the DIIIS4–S5 linker is known to interact with the hydrophobic isoleucine-phenylalanine-methionine-threonine (IFMT) amino acid sequence in the inactivation gate (Goldin, 2003; Smith and Goldin, 1997). Two recent studies characterized the biophysical consequences of two pathogenic variants located in the DIIIS4–S5 linker, I1327V and L1331V. As predicted, both variants impair channel inactivation, resulting in channels that are slower to transition from the open state to the inactive state (Barker et al., 2016; Patel et al., 2016). Additionally, amino acid substitutions have been identified at p.A1319 in the DIIIS4–S5 linker, as well as at the isoleucine (p.I1479), phenylalanine (p.F1480), and methionine (p.M1481) residues in the inactivation gate in epilepsy patients. A total of seven unique epilepsy variants have been observed within the SCN8A inactivation gate, including the recurrent p.E1483K inherited missense variant. Variants that alter the structure of the inactivation gate also have the potential to alter channel inactivation, producing hyperexcitable channels.
The C-terminal domain also plays a role in modulating fast inactivation of VGSCs. Specifically, the proximal half of the C-terminus contains six α-helices that are predicted to interact with the inactivation gate, as well as with calmodulin, a calcium-responsive protein known to modulate channel inactivation (Cormier et al., 2002; O’Brien and Meisler, 2013). The cluster of seven pathogenic SCN8A variants observed in the C-terminus primarily affect the fifth α-helix, which contains multiple residues known to be important for inactivation kinetics (Nguyen and Goldin, 2010). Wagnon et al. showed that substitutions at the positively charged p.R1872 residue within the fifth α-helix destabilizes interactions with the inactivation gate, producing hyperexcitable channels (Wagnon et al., 2016). One can hypothesize that other variants in this region of the C-terminus may have similar effects on the inactivation kinetics of the channel, including the inherited variant p.N1877S reported in this study. Alternatively, substitutions in this region of the C-terminus may disrupt interactions with proteins that normally regulate channel activity or trafficking (O’Brien and Meisler, 2013).
5. Conclusions
In summary, we report several novel epilepsy variants in SCN8A identified by gene panel analysis. As genetic testing technologies continue to improve and become more integrated into routine clinical care, we will undoubtedly find other pathogenic SCN8A variants in patients with epilepsy and other neurodevelopmental disorders. These additional patients will provide insight into the full phenotypic spectrum of SCN8A dysfunction and improve our ability to interpret new variants uncovered by diagnostic testing. Efforts are currently underway to launch an SCN8A variant registry (http://www.scn8a.net/), which will allow families, clinicians, and researchers to come together to advance our understanding of SCN8A and epilepsy.
Highlights.
Five SCN8A variants were identified in individuals with severe and mild epilepsy.
Variation in SCN8A may contribute to neurodevelopmental disorders without seizures.
Four distinct clusters of pathogenic variants were identified within the channel.
These clusters are located in regions of the channel important for inactivation.
Acknowledgments
We would like to thank the families for their participation. We are grateful for the assistance of Cynthia Freehauf in the collection of clinical data. This study was supported by funding from Children’s Healthcare of Atlanta to A.E., the Emory University Research Council to A.E. and J.J.A., and a training grant appointment (5T32GM008490-23) to K.M.B. Deep amplicon sequencing was performed by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand G, Collett-White F, Orsini A, Thomas S, Jayapal S, Trump N, Zaiwalla Z, Jayawant S. Autosomal dominant SCN8A mutation with an unusually mild phenotype. European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society. 2016 doi: 10.1016/j.ejpn.2016.04.015. [DOI] [PubMed] [Google Scholar]
- Barker BS, Ottolini M, Wagnon JL, Hollander RM, Meisler MH, Patel MK. The SCN8A encephalopathy mutation p.Ile1327Val displays elevated sensitivity to the anticonvulsant phenytoin. Epilepsia. 2016 doi: 10.1111/epi.13461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovic SF, Heron SE, Giordano L, Marini C, Guerrini R, Kaplan RE, Gambardella A, Steinlein OK, Grinton BE, Dean JT, Bordo L, Hodgson BL, Yamamoto T, Mulley JC, Zara F, Scheffer IE. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Annals of neurology. 2004;55:550–557. doi: 10.1002/ana.20029. [DOI] [PubMed] [Google Scholar]
- Blanchard MG, Willemsen MH, Walker JB, Dib-Hajj SD, Waxman SG, Jongmans MC, Kleefstra T, van de Warrenburg BP, Praamstra P, Nicolai J, Yntema HG, Bindels RJ, Meisler MH, Kamsteeg EJ. De novo gain-of-function and loss-of-function mutations of SCN8A in patients with intellectual disabilities and epilepsy. Journal of medical genetics. 2015;52:330–337. doi: 10.1136/jmedgenet-2014-102813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:5616–5620. doi: 10.1073/pnas.090034797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvill GL, Regan BM, Yendle SC, O’Roak BJ, Lozovaya N, Bruneau N, Burnashev N, Khan A, Cook J, Geraghty E, Sadleir LG, Turner SJ, Tsai MH, Webster R, Ouvrier R, Damiano JA, Berkovic SF, Shendure J, Hildebrand MS, Szepetowski P, Scheffer IE, Mefford HC. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nature genetics. 2013;45:1073–1076. doi: 10.1038/ng.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. American journal of human genetics. 2001;68:1327–1332. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormier JW, Rivolta I, Tateyama M, Yang AS, Kass RS. Secondary structure of the human cardiac Na+ channel C terminus: evidence for a role of helical structures in modulation of channel inactivation. The Journal of biological chemistry. 2002;277:9233–9241. doi: 10.1074/jbc.M110204200. [DOI] [PubMed] [Google Scholar]
- de Kovel CG, Meisler MH, Brilstra EH, van Berkestijn FM, van ‘t Slot R, van Lieshout S, Nijman IJ, O’Brien JE, Hammer MF, Estacion M, Waxman SG, Dib-Hajj SD, Koeleman BP. Characterization of a de novo SCN8A mutation in a patient with epileptic encephalopathy. Epilepsy research. 2014;108:1511–1518. doi: 10.1016/j.eplepsyres.2014.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi KC, Allen AS, Berkovic SF, Cossette P, Delanty N, Dlugos D, Eichler EE, Epstein MP, Glauser T, Goldstein DB, Han Y, Heinzen EL, Hitomi Y, Howell KB, Johnson MR, Kuzniecky R, Lowenstein DH, Lu YF, Madou MR, Marson AG, Mefford HC, Esmaeeli Nieh S, O’Brien TJ, Ottman R, Petrovski S, Poduri A, Ruzzo EK, Scheffer IE, Sherr EH, Yuskaitis CJ, Abou-Khalil B, Alldredge BK, Bautista JF, Berkovic SF, Boro A, Cascino GD, Consalvo D, Crumrine P, Devinsky O, Dlugos D, Epstein MP, Fiol M, Fountain NB, French J, Friedman D, Geller EB, Glauser T, Glynn S, Haut SR, Hayward J, Helmers SL, Joshi S, Kanner A, Kirsch HE, Knowlton RC, Kossoff EH, Kuperman R, Kuzniecky R, Lowenstein DH, McGuire SM, Motika PV, Novotny EJ, Ottman R, Paolicchi JM, Parent JM, Park K, Poduri A, Scheffer IE, Shellhaas RA, Sherr EH, Shih JJ, Singh R, Sirven J, Smith MC, Sullivan J, Lin Thio L, Venkat A, Vining EP, Von Allmen GK, Weisenberg JL, Widdess-Walsh P, Winawer MR Epilepsy Phenome/Genome P. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nature genetics. 2000;24:343–345. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- Estacion M, O’Brien JE, Conravey A, Hammer MF, Waxman SG, Dib-Hajj SD, Meisler MH. A novel de novo mutation of SCN8A (Nav1.6) with enhanced channel activation in a child with epileptic encephalopathy. Neurobiology of disease. 2014;69:117–123. doi: 10.1016/j.nbd.2014.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EV, Kullmann DM, Schorge S. Alternative splicing modulates inactivation of type 1 voltage-gated sodium channels by toggling an amino acid in the first S3-S4 linker. The Journal of biological chemistry. 2011;286:36700–36708. doi: 10.1074/jbc.M111.250225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadzicki D, Docker D, Schubach M, Menzel M, Schmorl B, Stellmer F, Biskup S, Bartholdi D. Expanding the phenotype of a recurrent de novo variant in PACS1 causing intellectual disability. Clinical genetics. 2015;88:300–302. doi: 10.1111/cge.12544. [DOI] [PubMed] [Google Scholar]
- Gardella E, Becker F, Moller RS, Schubert J, Lemke JR, Larsen LH, Eiberg H, Nothnagel M, Thiele H, Altmuller J, Syrbe S, Merkenschlager A, Bast T, Steinhoff B, Nurnberg P, Mang Y, Bakke Moller L, Gellert P, Heron SE, Dibbens LM, Weckhuysen S, Dahl HA, Biskup S, Tommerup N, Hjalgrim H, Lerche H, Beniczky S, Weber YG. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Annals of neurology. 2016;79:428–436. doi: 10.1002/ana.24580. [DOI] [PubMed] [Google Scholar]
- Gazina EV, Leaw BT, Richards KL, Wimmer VC, Kim TH, Aumann TD, Featherby TJ, Churilov L, Hammond VE, Reid CA, Petrou S. ‘Neonatal’ Nav1.2 reduces neuronal excitability and affects seizure susceptibility and behaviour. Human molecular genetics. 2015;24:1457–1468. doi: 10.1093/hmg/ddu562. [DOI] [PubMed] [Google Scholar]
- Gazina EV, Richards KL, Mokhtar MB, Thomas EA, Reid CA, Petrou S. Differential expression of exon 5 splice variants of sodium channel alpha subunit mRNAs in the developing mouse brain. Neuroscience. 2010;166:195–200. doi: 10.1016/j.neuroscience.2009.12.011. [DOI] [PubMed] [Google Scholar]
- Goldin AL. Mechanisms of sodium channel inactivation. Current opinion in neurobiology. 2003;13:284–290. doi: 10.1016/s0959-4388(03)00065-5. [DOI] [PubMed] [Google Scholar]
- Helbig KL, Farwell Hagman KD, Shinde DN, Mroske C, Powis Z, Li S, Tang S, Helbig I. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genetics in medicine : official journal of the American College of Medical Genetics. 2016 doi: 10.1038/gim.2015.186. [DOI] [PubMed] [Google Scholar]
- Howell KB, McMahon JM, Carvill GL, Tambunan D, Mackay MT, Rodriguez-Casero V, Webster R, Clark D, Freeman JL, Calvert S, Olson HE, Mandelstam S, Poduri A, Mefford HC, Harvey AS, Scheffer IE. SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology. 2015;85:958–966. doi: 10.1212/WNL.0000000000001926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodera H, Kato M, Nord AS, Walsh T, Lee M, Yamanaka G, Tohyama J, Nakamura K, Nakagawa E, Ikeda T, Ben-Zeev B, Lev D, Lerman-Sagie T, Straussberg R, Tanabe S, Ueda K, Amamoto M, Ohta S, Nonoda Y, Nishiyama K, Tsurusaki Y, Nakashima M, Miyake N, Hayasaka K, King MC, Matsumoto N, Saitsu H. Targeted capture and sequencing for detection of mutations causing early onset epileptic encephalopathy. Epilepsia. 2013;54:1262–1269. doi: 10.1111/epi.12203. [DOI] [PubMed] [Google Scholar]
- Larsen J, Carvill GL, Gardella E, Kluger G, Schmiedel G, Barisic N, Depienne C, Brilstra E, Mang Y, Nielsen JE, Kirkpatrick M, Goudie D, Goldman R, Jahn JA, Jepsen B, Gill D, Docker M, Biskup S, McMahon JM, Koeleman B, Harris M, Braun K, de Kovel CG, Marini C, Specchio N, Djemie T, Weckhuysen S, Tommerup N, Troncoso M, Troncoso L, Bevot A, Wolff M, Hjalgrim H, Guerrini R, Scheffer IE, Mefford HC, Moller RS double daggerOn behalf of the Euro, E.R.E.S.C.C.R.P., Euro, E.R.E.S.C.C.R.P. The phenotypic spectrum of SCN8A encephalopathy. Neurology. 2015;84:480–489. doi: 10.1212/WNL.0000000000001211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Cai T, Jiang Y, Chen H, He X, Chen C, Li X, Shao Q, Ran X, Li Z, Xia K, Liu C, Sun ZS, Wu J. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Molecular psychiatry. 2015 doi: 10.1038/mp.2015.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinson CD, Tanaka BS, Lamar T, Goldin AL, Escayg A. Role of the hippocampus in Nav1.6 (Scn8a) mediated seizure resistance. Neurobiology of disease. 2014;68:16–25. doi: 10.1016/j.nbd.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Human molecular genetics. 2007;16:2892–2899. doi: 10.1093/hmg/ddm248. [DOI] [PubMed] [Google Scholar]
- McMichael G, Bainbridge MN, Haan E, Corbett M, Gardner A, Thompson S, van Bon BW, van Eyk CL, Broadbent J, Reynolds C, O’Callaghan ME, Nguyen LS, Adelson DL, Russo R, Jhangiani S, Doddapaneni H, Muzny DM, Gibbs RA, Gecz J, MacLennan AH. Whole-exome sequencing points to considerable genetic heterogeneity of cerebral palsy. Molecular psychiatry. 2015;20:176–182. doi: 10.1038/mp.2014.189. [DOI] [PubMed] [Google Scholar]
- Meisler MH, Helman G, Hammer MF, Fureman BE, Gaillard WD, Goldin AL, Hirose S, Ishii A, Kroner BL, Lossin C, Mefford HC, Parent JM, Patel M, Schreiber J, Stewart R, Whittemore V, Wilcox K, Wagnon JL, Pearl PL, Vanderver A, Scheffer IE. SCN8A encephalopathy: Research progress and prospects. Epilepsia. 2016 doi: 10.1111/epi.13422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, Kearney J, Escayg A, MacDonald BT, Sprunger LK. Sodium channels and neurological disease: insights from Scn8a mutations in the mouse. The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry. 2001;7:136–145. doi: 10.1177/107385840100700208. [DOI] [PubMed] [Google Scholar]
- Mercimek-Mahmutoglu S, Patel J, Cordeiro D, Hewson S, Callen D, Donner EJ, Hahn CD, Kannu P, Kobayashi J, Minassian BA, Moharir M, Siriwardena K, Weiss SK, Weksberg R, Snead OC., 3rd Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia. 2015;56:707–716. doi: 10.1111/epi.12954. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Kato M, Osaka H, Yamashita S, Nakagawa E, Haginoya K, Tohyama J, Okuda M, Wada T, Shimakawa S, Imai K, Takeshita S, Ishiwata H, Lev D, Lerman-Sagie T, Cervantes-Barragan DE, Villarroel CE, Ohfu M, Writzl K, Gnidovec Strazisar B, Hirabayashi S, Chitayat D, Myles Reid D, Nishiyama K, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Hayasaka K, Matsumoto N, Saitsu H. Clinical spectrum of SCN2A mutations expanding to Ohtahara syndrome. Neurology. 2013;81:992–998. doi: 10.1212/WNL.0b013e3182a43e57. [DOI] [PubMed] [Google Scholar]
- Nguyen HM, Goldin AL. Sodium channel carboxyl-terminal residue regulates fast inactivation. The Journal of biological chemistry. 2010;285:9077–9089. doi: 10.1074/jbc.M109.054940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JE, Meisler MH. Sodium channel SCN8A (Nav1.6): properties and de novo mutations in epileptic encephalopathy and intellectual disability. Frontiers in genetics. 2013;4:213. doi: 10.3389/fgene.2013.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, Rieder MJ, Nickerson DA, Bernier R, Fisher SE, Shendure J, Eichler EE. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nature genetics. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba C, Kato M, Takahashi S, Lerman-Sagie T, Lev D, Terashima H, Kubota M, Kawawaki H, Matsufuji M, Kojima Y, Tateno A, Goldberg-Stern H, Straussberg R, Marom D, Leshinsky-Silver E, Nakashima M, Nishiyama K, Tsurusaki Y, Miyake N, Tanaka F, Matsumoto N, Saitsu H. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia. 2014;55:994–1000. doi: 10.1111/epi.12668. [DOI] [PubMed] [Google Scholar]
- Oliva M, Berkovic SF, Petrou S. Sodium channels and the neurobiology of epilepsy. Epilepsia. 2012;53:1849–1859. doi: 10.1111/j.1528-1167.2012.03631.x. [DOI] [PubMed] [Google Scholar]
- Patel RR, Barbosa C, Brustovetsky T, Brustovetsky N, Cummins TR. Aberrant epilepsy-associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain : a journal of neurology. 2016 doi: 10.1093/brain/aww129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, Albrecht B, Bartholdi D, Beygo J, Di Donato N, Dufke A, Cremer K, Hempel M, Horn D, Hoyer J, Joset P, Röpke A, Moog U, Riess A, Thiel CT, Tzschach A, Wiesener A, Wohlleber E, Zweier C, Ekici AB, Zink AM, Rump A, Meisinger C, Grallert H, Sticht H, Schenck A, Engels H, Rappold G, Schröck E, Wieacker P, Riess O, Meitinger T, Reis A, Strom TM. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. The Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- Raymond CK, Castle J, Garrett-Engele P, Armour CD, Kan Z, Tsinoremas N, Johnson JM. Expression of alternatively spliced sodium channel alpha-subunit genes. Unique splicing patterns are observed in dorsal root ganglia. The Journal of biological chemistry. 2004;279:46234–46241. doi: 10.1074/jbc.M406387200. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, Walker MF, Ober GT, Teran NA, Song Y, El-Fishawy P, Murtha RC, Choi M, Overton JD, Bjornson RD, Carriero NJ, Meyer KA, Bilguvar K, Mane SM, Sestan N, Lifton RP, Gunel M, Roeder K, Geschwind DH, Devlin B, State MW. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Goldin AL. Interaction between the sodium channel inactivation linker and domain III S4-S5. Biophysical journal. 1997;73:1885–1895. doi: 10.1016/S0006-3495(97)78219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Mullegama SV, Rosenfeld JA, van Bon BW, Shen Y, Repnikova EA, Gastier-Foster J, Thrush DL, Kathiresan S, Ruderfer DM, Chiang C, Hanscom C, Ernst C, Lindgren AM, Morton CC, An Y, Astbury C, Brueton LA, Lichtenbelt KD, Ades LC, Fichera M, Romano C, Innis JW, Williams CA, Bartholomew D, Van Allen MI, Parikh A, Zhang L, Wu BL, Pyatt RE, Schwartz S, Shaffer LG, de Vries BB, Gusella JF, Elsea SH. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. American journal of human genetics. 2011;89:551–563. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau MM, Dalton JC, Day JW, Ranum LP, Meisler MH. Heterozygosity for a protein truncation mutation of sodium channel SCN8A in a patient with cerebellar atrophy, ataxia, and mental retardation. Journal of medical genetics. 2006;43:527–530. doi: 10.1136/jmg.2005.035667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeramah KR, O’Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, Talwar D, Girirajan S, Eichler EE, Restifo LL, Erickson RP, Hammer MF. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. American journal of human genetics. 2012;90:502–510. doi: 10.1016/j.ajhg.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Barker BS, Hounshell JA, Haaxma CA, Shealy A, Moss T, Parikh S, Messer RD, Patel MK, Meisler MH. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Annals of clinical and translational neurology. 2016;3:114–123. doi: 10.1002/acn3.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Korn MJ, Parent R, Tarpey TA, Jones JM, Hammer MF, Murphy GG, Parent JM, Meisler MH. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Human molecular genetics. 2015;24:506–515. doi: 10.1093/hmg/ddu470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Meisler MH. Recurrent and Non-Recurrent Mutations of SCN8A in Epileptic Encephalopathy. Frontiers in neurology. 2015;6:104. doi: 10.3389/fneur.2015.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Thomas EA, Jenkins M, Gazina EV, Chiu C, Heron SE, Mulley JC, Scheffer IE, Berkovic SF, Petrou S. A childhood epilepsy mutation reveals a role for developmentally regulated splicing of a sodium channel. Molecular and cellular neurosciences. 2007;35:292–301. doi: 10.1016/j.mcn.2007.03.003. [DOI] [PubMed] [Google Scholar]