

In this review, Dorn et al. describe the regulatory circuitry and downstream events involved in mitochondrial biogenesis and its coordination with mitochondrial dynamics in developing and diseased hearts.
Keywords: mitochondria, biogenesis, mitophagy, mitochondrial dynamics
Abstract
The mitochondrion is a complex organelle that serves essential roles in energy transduction, ATP production, and a myriad of cellular signaling events. A finely tuned regulatory network orchestrates the biogenesis, maintenance, and turnover of mitochondria. The high-capacity mitochondrial system in the heart is regulated in a dynamic way to generate and consume enormous amounts of ATP in order to support the constant pumping function in the context of changing energy demands. This review describes the regulatory circuitry and downstream events involved in mitochondrial biogenesis and its coordination with mitochondrial dynamics in developing and diseased hearts.
The adult human heart generates and consumes kilogram quantities of ATP daily to support normal pump function. Mitochondrial oxidative phosphorylation (OXPHOS) is responsible for nearly all of the ATP production (>95%) in adult mammalian hearts (Ashrafian et al. 2007). The creatine phosphate shuttle system delivers the high-energy phosphate groups from the site of production in the mitochondria to myofibrils to regenerate ATP consumed during contraction. As such, the heart requires an enormous mitochondrial biogenic capacity to support the high demand for ATP production. In fact, >40% of the cytoplasmic space (Hom and Sheu 2009) in adult cardiac myocytes is occupied by mitochondria densely packed between sarcomeres, around the nucleus, and in the subsarcolemma. The development and maturation of this specialized high-capacity mitochondrial system in the heart occur largely during the perinatal and postnatal developmental stages. The process begins with a major surge in mitochondrial biogenesis at birth. Following this biogenic surge, there is a period of maturation that involves a dramatic increase in dynamics (mitophagy, fusion, and fission), leading to redistribution and dense packing of the specialized mature mitochondria along myofibrils. This cellular architecture facilitates the transfer of high-energy phosphates between the mitochondria and the contractile apparatus. The resultant mature mitochondrial system is capable of high-capacity oxidation of fuels such as fatty acid, the predominant fuel substrate for the adult heart. This review focuses on current knowledge of the regulatory circuitry and mechanisms involved in the maturation and maintenance of this specialized high-capacity mitochondrial system in developing and adult mammalian hearts.
Mitochondrial biogenesis: circuitry and regulatory mechanisms
Mitochondrial genomic and energy transduction machinery
The mitochondrion is a double-membrane organelle with an ion-permeable inner membrane and an outer membrane permeable to factors <5 kDa (Balaban 1990). This complex membrane structure enables ATP generation via OXPHOS. An electrochemical gradient established across the inner membrane drives OXPHOS and ATP synthesis. This gradient involves electrons donated from reduced forms of nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) generated by oxidation of acetyl-CoA via the tricarboxylic acid (TCA) cycle. The acetyl-CoA is generated by oxidation of energy substrates, chiefly fatty acids and glucose–pyruvate in normal adult hearts (Stanley et al. 2005). The electrons are accepted and transferred across components of the electron transport chain (ETC) in the inner mitochondrial membrane. The transfer of electrons along the ETC is coupled to the transport of protons across the inner membrane, establishing the electrochemical gradient that drives ATP synthesis.
The mitochondrion is a unique organelle in that it contains its own self-replicating genome. The mitochondrial DNA (mtDNA) encodes 13 essential components of the ETC as well as all rRNAs and tRNAs necessary for translation of the mtDNA-encoded proteins (Scarpulla et al. 2012). However, the vast majority of mitochondrial proteins (>1000) (Pagliarini et al. 2008; Lotz et al. 2014) are encoded by the nuclear genome. Therefore, the biogenesis of mitochondria requires exquisite coordination of both mitochondrial and nuclear genomes. The control mechanisms for the orchestrated control of mitochondrial biogenesis are achieved largely through factors encoded by the nuclear genome, as described below. Indeed, all of the machinery needed for replication and transcription of the mitochondrial genome is contained within the >1000 nuclear-encoded mitochondrial proteins.
Biogenic machinery: transcriptional networks
A complex transcriptional network orchestrates nuclear and mitochondrial genome transcription and replication to conduct robust and dynamic mitochondrial biogenic responses in the heart. This system must coordinate both genomes during development and in response to physiological cues when there are changes in fuel substrate availability or energetic demands. A key breakthrough in our understanding of the transcriptional control of mitochondrial biogenesis and maturation came with the identification of the transcriptional coregulator peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α (PGC-1α). PGC-1α was first identified in brown adipose as a direct coactivator of PPARγ, a master regulator of adipogenesis (Puigserver et al. 1998). PGC-1α is a member of a family of transcriptional coregulators, including the closely related PGC-1β and a more distant member, PGC-1-related coactivator (PRC) (Andersson and Scarpulla 2001; Kressler et al. 2002; Lin et al. 2002). Gain- and loss-of-function studies have revealed a critical role for the PGC-1 coactivators in driving mitochondrial biogenesis and function in the heart and other mitochondrial-rich organs such as brown adipose tissue and skeletal muscle. Cardiac-specific transgenic overexpression of PGC-1α in mice results in an exuberant mitochondrial biogenic response during the postnatal period together with increased expression of nuclear-encoded mitochondrial genes (Lehman et al. 2000). Genetic deletion of either PGC-1α or PGC-1β in mice does not lead to overt abnormalities in mitochondrial function or biogenesis under basal conditions, indicating functional redundancy. However, loss of PGC-1α or PGC-1β accelerates cardiac dysfunction following the stress of pressure overload (Arany et al. 2006). Germline deletion of both PGC-1α and PGC-1β evokes perinatal lethal heart failure caused by a complete lack of cardiac mitochondrial biogenesis (Fig. 1; Lai et al. 2008). Mice lacking PRC exhibit an embryonic-lethal phenotype, dying shortly after implantation. These latter results support a critical and novel role for PRC in embryonic development (He et al. 2012). Taken together, these results indicate that PGC-1 signaling is both sufficient and necessary for cardiac mitochondrial biogenesis at birth and that PGC-1α and PGC-1β have overlapping functions.
Figure 1.

Cardiac mitochondrial phenotypes of developmental stage-specific PGC-1 knockout mice. Electron micrographs illustrate the altered mitochondrial ultrastructure and biogenic response resultant from developmental stage- and cardiac-specific PGC-1α/β gene targeting “knockouts” (α/β−/−) (bottom row) compared with wild-type or PGC-1α−/− (adult) age-matched controls (top row). (Bottom, left) Germline disruption of PGC-1α/β genes results in a perinatal arrest of biogenesis with a profound reduction in mitochondrial content, as characterized by small numbers of immature mitochondria on day 1 following birth. (Bottom, middle) Deletion of cardiac PGC-1α/β genes in the postnatal period (using a muscle creatine kinase-driven Cre recombinase) impairs mitochondrial fusion and fission, as reflected by fragmented, elongated, and “donut”-shaped mitochondria. The micrographs were taken of cardiac ventricles at postnatal day 28. (Bottom, right) Inducible deletion of PGC-1 in adult hearts (12 wk of age) does not significantly affect mitochondrial density but results in a subset of abnormal mitochondria with collapsed cristae (white arrow), reminiscent of the phospholipid abnormalities seen with human Barth syndrome. Bars: germline micrographs, 0.5 μm; postnatal and adult micrographs, 1.0 μm. (FAO) Fatty acid oxidation.
The mouse phenotypes resulting from developmental stage-specific targeting of PGC-1β on a generalized PGC-1α-deficient background in mice has provided important insight into the roles of the coregulators in postnatal and adult mammalian hearts. Targeting of the PGC-1 coactivators after the perinatal biogenic response results in a progressive postnatal cardiomyopathy associated with dramatic mitochondrial morphological derangements indicative of derangements in mitochondrial fusion and fission (Fig. 1; Martin et al. 2014). These latter results demonstrate the importance of PGC-1 signaling for postnatal mitochondrial maturation, including a critical role for mitochondrial dynamics during growth of postnatal hearts. As described below, mitophagy is also required for this maturation process and is therefore included as a component of the process of mitochondrial dynamics.
Surprisingly, and in contrast to the germline and postnatal knockout phenotypes, inducible deletion of PGC-1 in adult hearts in mice does not compromise normal cardiac function or result in overt abnormalities of mitochondrial structure. However, adult PGC-1α/β-deficient mice exhibit global down-regulation of transcripts encoding components of the fatty acid oxidation (FAO), TCA, and ETC/OXPHOS pathways together with decreased respiratory capacity (Martin et al. 2014). These findings as well as observations by others (Chen et al. 2011) indicate that rates of mitochondrial fission and fusion (and mitochondrial turnover) are much lower in normal adult hearts compared with during postnatal developmental stages. Interestingly, a subset of mitochondria in adult PGC-1 double-knockout mice displayed ultrastructural mitochondrial cristae abnormalities similar to what is observed in Barth syndrome, a congenital disease caused by altered cardiolipin biosynthesis (Lai et al. 2014b). These latter results implicate PGC-1 in mitochondrial membrane lipid biosynthesis. Taken together with the results of the perinatal and postnatal PGC-1-deficient phenotypes, these studies have identified developmental stage-specific roles for the PGC-1 coactivators in the heart (Fig. 1).
The PGC-1 coactivators exert their function through direct interactions with target transcription factor effectors, allowing coordinate control of the various pathways involved in building new mitochondria (Fig. 2; Vega et al. 2015). Most of the work defining such interactions has focused on PGC-1α. PGC-1α enhances transcription by interacting directly with members of the nuclear receptor superfamily via specific LXXLL recognition domains, recruiting molecules that mediate chromatin remodeling via histone acetylation, and interacting with the TRAP/DRIP complex to recruit RNA polymerase II (Ge et al. 2002). Effector transcription factors within this cascade include members of the PPAR, estrogen-related receptor (ERR), and nuclear respiratory factor-1 (NRF-1) transcription factor families. As shown in Figure 2, these transcription factors control and regulate distinct, but at times overlapping, aspects of mitochondrial energy metabolism, respiration, and biogenesis.
Figure 2.
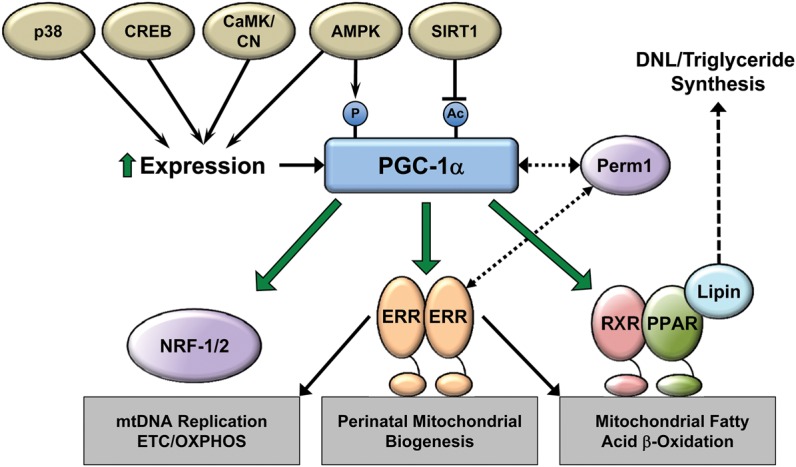
The PGC-1α transcriptional regulatory cascade: upstream inputs and downstream targets. PGC-1α expression and activity are modulated by various upstream signaling pathways responsive to physiologic and metabolic stimuli to control mitochondrial biogenesis and function. As shown, PGC-1α interacts directly with and coactivates multiple DNA-binding transcription factors to control virtually all aspects of biogenesis, dynamics, and maintenance of mitochondrial protein levels. (CREB) cAMP response element-binding protein, (CaMK) calmodulin-dependent kinase, (CN) calcineurin, (AMPK) AMP-activated kinase, (SIRT1) sirtuin 1, (Perm1) PGC-1 and ERR regulator in muscle 1, (RXR) retinoid X receptor.
NRF-1 and NRF-2 (GABP) regulate expression of virtually every complex in the ETC (Scarpulla 2008; Satoh et al. 2013). In addition, the PGC-1/NRF-1 interaction activates downstream factors involved in mtDNA replication. Specifically, NRF-1 activates transcription of genes encoding factors that mediate replication and transcription of the mitochondrial genome, including TFAM and TFB2M (Gleyzer et al. 2005). Consistent with these roles, genetic loss of either NRF-1 or NRF-2 results in embryonic lethality with reduced mtDNA content and ETC activity, underscoring the necessary role for each of these factors (Huo and Scarpulla 2001; Ristevski et al. 2004).
The ERR family of nuclear receptors serves a central function in the PGC-1 regulatory circuitry (Fig. 2). ERRα, ERRβ, and ERRγ have significant homology with classic estrogen receptors in the ligand-binding domain but do not bind estrogen or any known endogenous ligand (“orphan” nuclear receptors). The first evidence supporting a role for ERRα in the regulation of mitochondrial metabolism was the discovery that it regulates transcription of the gene encoding medium chain acyl-CoA dehydrogenase (MCAD), an enzyme that catalyzes the first step in mitochondrial FAO (Sladek et al. 1997; Vega and Kelly 1997). Broader links to PGC-1 and mitochondrial biogenesis were made when it was shown that PGC-1α interacts with ERRα via the results of a yeast two-hybrid screen (Huss et al. 2002; Schreiber et al. 2004). In cooperation with PGC-1α, ERRα has since been shown to regulate genes in virtually every pathway of mitochondrial energy transduction and ATP synthesis, including FAO, the TCA cycle, and ETC/OXPHOS (Huss et al. 2004). Genome-wide chromatin immunoprecipitation (ChIP) surveys confirmed direct binding of ERRα and ERRγ to the promoter regions of many nuclear-encoded mitochondrial genes (Dufour et al. 2007). Generalized ERRα knockout mice are fertile and viable but are susceptible to cardiac dysfunction in response to pressure overload and have reduced ATP synthesis rates following ischemic insult (Huss et al. 2007). ERRγ knockout mice die shortly after birth of heart failure with an inability to shift to oxidative metabolism (Alaynick et al. 2007). These latter results point to a critical role by ERRγ in the postnatal energy metabolic maturation of developing hearts. Recently, it has been shown that combined cardiac-specific deletion of ERRα/ERRγ displays premature lethality with reduced expression of genes critical for mitochondrial energy production (Wang et al. 2015). The role of the third member of the family, ERRβ, in cardiac mitochondrial function and energy production is not known. Notably, ERRα and ERRγ have also been shown to regulate contractile and calcium-handling gene expression in the heart and skeletal muscle, suggesting functions in coordinate control of mitochondrial energy production and contractile function in striated muscle (Dufour et al. 2007; Gan et al. 2013).
The PPAR family of nuclear receptor transcription factors can also serve as effectors of the PGC-1 coactivators. The PPARs (PPARα, PPARδ [also known as PPARβ], and PPARγ) were originally identified as regulators of peroxisomal β-oxidation (Issemann and Green 1990). PPARs are now known to regulate genes involved in mitochondrial FAO and many other cellular lipid metabolic pathways (van der Meer et al. 2010; Scarpulla et al. 2012; Kersten 2014; McMullen et al. 2014). Given that fatty acids are the preferred fuel substrate in normal adult hearts, accounting for 60%–90% of ATP production, the PPARs serve a critical role in the biogenesis of mitochondria specialized for high-capacity FAO in the heart. Target genes for PPARα in the heart include enzymes and proteins involved in fatty acid transport, activation, and mitochondrial FAO (Gulick et al. 1994; Brandt et al. 1998; Mascaro et al. 1998). The function of PPARs as regulators of cardiac myocyte fatty acid metabolism has been validated in vivo by PPARα loss of function and transgenic overexpression in mice (Aoyama et al. 1998; Djouadi et al. 1999; Leone et al. 1999; Watanabe et al. 2000; Finck et al. 2002). PPARs have large, hydrophobic ligand-binding domains that accommodate a variety of endogenous fatty acids and eicosanoids as activating ligands. In this way, PPARs serve as metabolic sensors to match fuel substrate availability and delivery with high FAO capacity. Although all PPARs bind as heterodimers to their cognate DNA elements with the retinoid X receptor (RXR), they may also interact with other transcription factors. For instance, recent work has shown that the Kruppel-like factor 15 (KLF15) interacts directly with PPARα in the heart to regulate genes involved in FAO (Prosdocimo et al. 2014, 2015).
In addition to the PGC-1 regulatory circuit, several other transcription factors have been shown to contribute to mitochondrial biogenesis. Such factors likely function independently of the PGC-1 coactivators in distinct physiologic or pathophysiologic contexts. The c-Myc transcription factor has been shown to induce mitochondrial function and activate NRF-1 and TFAM (Li et al. 2005; Kim et al. 2008). In the heart, interestingly, c-Myc triggers increased reliance on glucose and decreased FAO during times of growth or ischemia, a possible adaptive response to stress (Ahuja et al. 2010). In support of this latter role, c-Myc expression increases with pressure overload and improves recovery following an ischemic insult (Ahuja et al. 2010). These results suggest that c-Myc plays a unique role in the control of mitochondrial biogenesis and function when canonical pathways (PGC-1/ERR and PGC-1/PPAR) are not active, such as occurs during fetal periods or hypertrophic growth when the heart relies to a greater extent on glucose as a fuel substrate.
Regulatory inputs into mitochondrial biogenesis via regulation of PGC-1 coactivators
PGC-1α receives inputs from multiple pathways to control its expression and activity in diverse developmental and physiological contexts. As shown in Figure 2, PGC-1α expression is highly induced by exposure to cold and exercise, mediated largely through β-adrenergic stimulation and cAMP/CREB (cAMP response element-binding protein) signaling (Wu et al. 1999; Baar et al. 2002; Terada et al. 2002). In skeletal muscle, p38 MAPK also stimulates PGC-1α gene expression following a single bout of exercise mediated through the ATF2 transcription factor (Akimoto et al. 2005). Activation of AMPK (AMP-activated kinase) following acute exercise may also increase PGC-1α expression in skeletal muscle (Little et al. 2010). Activation of the calmodulin-dependent kinase and calcineurin by calcium signaling provides an additional mechanism to increase PGC-1α expression (Czubryt et al. 2003; Schaeffer et al. 2004). Finally, in the heart, the proviral integration site for Moloney murine leukemia virus (PIM) kinases has been shown to regulate PGC-1 expression and capacity for mitochondrial energy production. Interestingly, genetic deletion of all three Pim kinases (Pim-1, Pim-2, and Pim-3) results in a cardiac senescence phenotype with marked reduction of PGC-1α and PGC-1β concomitant with mitochondrial defects and decreased ATP levels (Din et al. 2014).
PGC-1α activity is also regulated by post-translational modifications, including phosphorylation and acetylation (Fig. 2). There is some evidence that phosphorylation by AMP kinase activates PGC-1α, providing a link between cellular energy status and mitochondrial biogenesis (Jager et al. 2007). PGC-1α has been shown to be acetylated at multiple lysine residues and is a substrate for the NAD+-dependent deacetylase sirtuin 1 (SIRT1) (Gerhart-Hines et al. 2007; Coste et al. 2008; Canto et al. 2009). As NAD+ levels are also regulated by AMPK, activation of PGC-1α through deacetylation by SIRT1 potentially creates a link between energy status, redox state, and mitochondrial function (Canto et al. 2009).
PGC-1 coactivators have been shown to cooperate with other factors to promote mitochondrial biogenesis and function (Fig. 2). The PGC-1 and ERR regulator in muscle 1 (Perm1) was recently shown to be required for PGC-1-induced mitochondrial biogenesis (Cho et al. 2013). Perm1 expression is enriched in skeletal muscle, heart, and brown adipose and regulates the expression of certain PGC-1 and ERR target genes. Perm1 itself is activated by ERR/PGC-1, providing a feed-forward mechanism to promote mitochondrial biogenesis. A similar type of regulatory loop is active in melanocytes, where PGC-1α activates expression of the microphthalmia-associated transcription factor (MITF) to promote the tanning response (Shoag et al. 2013). MITF also directly activates PGC-1α expression to increase oxidative capacity to complete this feed-forward regulatory loop (Haq et al. 2013). This regulatory loop has been described in a subset of melanomas (Vazquez et al. 2013). Finally, interactions between PGC-1 and the phosphatidate phosphatase lipin 1 have been shown to modulate PGC-1 activity. Lipins were first discovered as enzymes that catalyze the formation of diacylglycerol in the glycerolipid biosynthesis pathway (Csaki and Reue 2010). However, lipin 1 also has a nuclear function by which it physically interacts with PGC-1α and PPARα to activate FAO gene expression (Finck et al. 2006). In the heart, lipin 1 expression is activated by ERR/PGC-1α forming a feed-forward loop to modulate cardiac metabolism (Mitra et al. 2011).
Mitophagy
It seems evident that homeostasis in mitochondrial mass and tissue energetic demands requires biogenic synthesis of new mitochondrial components to be spatiotemporally matched with the removal of old or damaged mitochondria. The most completely understood mechanism for targeted removal of damaged mitochondria is through the PINK1 (PTEN-induced kinase 1)–Parkin mitophagy pathway (Fig. 3). As its name suggests, mitophagy describes consumption of whole or fragmented mitochondria by an autophagosome and transfer of the mitochondrial cargo to degradative lysosomes. Details of how PINK1 and Parkin interact were initially defined in studies using cultured cells and Drosophila skeletal muscle and have recently been reviewed in detail (Durcan and Fon 2015; Shirihai et al. 2015). Briefly, the nuclear-encoded kinase PINK1 is actively imported into mitochondria and, under normal conditions, undergoes rapid proteolytic degradation. Thus, PINK1 mRNA levels tend to be high, but protein levels are low; this is the case in normal mouse hearts (Song et al. 2015a). Mitochondrial dysfunction, measured experimentally as dissipation of the normal inner membrane electrochemical gradient (depolarization) or induction of the mitochondrial unfolded protein response (mtUPR), disrupts normal PINK1 proteolysis. Thus, PINK1 accumulates specifically in damaged mitochondria, where it phosphorylates a number of substrates that directly and indirectly attract Parkin to the organelle. Parkin is an E3 ubiquitin ligase that ubiquitinates at least 100 different mitochondrial outer membrane proteins upon PINK1-mediated mitochondrial translocation, thus targeting the organelle for docking to and engulfment by an autophagosome (Fig. 3).
Figure 3.
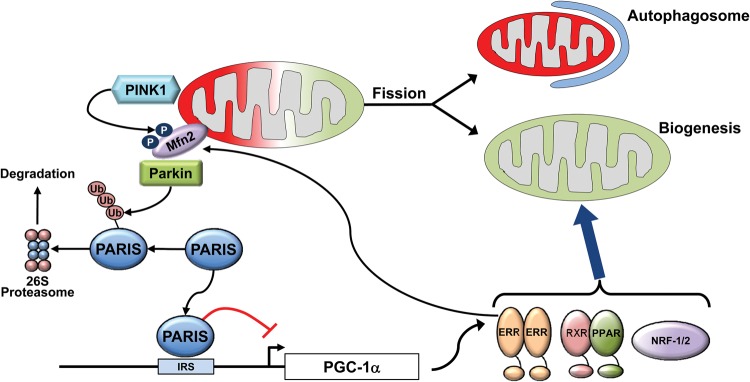
Coordinate control of mitophagy and mitochondrial biogenesis. Mitophagy and biogenesis are coordinately regulated to replace damaged mitochondria during periods of high mitochondrial turnover such as in the developing heart. Parkin is an E3 ubiquitin ligase recruited to the mitochondria through interaction with phosphorylated mitofusin 2 (Mfn2). Ubiquitination of outer mitochondrial membrane proteins by Parkin triggers partial or total engulfment by the autophagosome. Among Parkin's substrates is PARIS (Parkin-interacting substrate), a molecule that also serves as a transcriptional repressor of PGC-1α and may serve to coordinate mitophagy with biogenesis. Ubiquitination of PARIS and subsequent degradation serve to activate PGC-1α expression and the biogenic response. (IRS) Insulin response sequence.
Numerous loss-of-function PINK1 and Parkin mutations have been linked to autosomal recessive Parkinson's disease (Pickrell and Youle 2015), and it has been widely accepted that degeneration of dopaminergic neurons in Parkinson's disease is the consequence of interrupted mitophagy; i.e., a primary problem of mitochondrial quality control. The seminal observation underlying this pathogenic concept was that Parkin overexpression rescued mitochondrial defects in Drosophila PINK1 mutants (Clark et al. 2006; Park et al. 2006). However, PINK1 may not be necessary for Parkin translocation during mitophagy in mouse cardiac myocytes (Kubli et al. 2015), suggesting either that poorly described mitophagic mechanisms distinct from the canonical PINK1–Parkin pathway compensate for the absence of PINK1 or that mitophagy is not the only process that is rescued by overexpressing Parkin in PINK1-null flies. In other words, a rigorous interpretation of the fly data strongly suggests that Parkin can play central roles in biological processes other than and in addition to mitophagy. Indeed, there is little direct evidence that the PINK1–Parkin interaction mediates targeted mitophagy of damaged mitochondria in the dopaminergic neurons that are affected in Parkinson's disease (Scarffe et al. 2014), and germline Parkin knockout mice do not develop characteristic pathological features of Parkinson's disease (Lee et al. 2012b).
It has been speculated that Parkin and mitophagy play different roles in mitotic cells, such as the cultured fibroblasts that provided the foundation for our mechanistic understanding of mitophagy signaling, compared with nonmitotic terminally differentiated neurons (Scarffe et al. 2014). Like neurons, adult cardiomyocytes are post-mitotic and terminally differentiated, and neither germline nor cardiomyocyte-specific Parkin deletion significantly impacts unstressed adult hearts (Kubli et al. 2013b; Song et al. 2015a). Indeed, Parkin is present at such low abundance in normal adult mouse hearts as to be almost undetectable at both the mRNA and protein levels (Song et al. 2015a). Finally, there is direct evidence for the existence of Parkin-independent mitophagy mechanisms in neonatal and adult mouse hearts (Kageyama et al. 2014; Song et al. 2014). Thus, it is possible that nonmitophagic effects of Parkin, such as regulating mitochondrial biogenesis, represent more important housekeeping functions in normally functioning post-mitotic tissues such as the heart and brain than does mitochondrial quality control.
Orchestration of mitochondrial biogenesis and dynamics
In adult hearts, the rate of mitochondrial turnover is only approximately one-third of that of mitochondria in livers; i.e., approximately every 2 wk (Chen et al. 2011; Lau et al. 2015). Because measuring mitochondrial turnover as the absolute rate of component biosynthesis and mitochondrial removal is difficult in vivo, changes in mitochondrial biogenesis are often inferred from modulation of the characteristic PGC-1-directed cardiac gene expression program (Lai et al. 2008; Vega et al. 2015). By this metric, cardiomyocyte-specific Parkin deficiency (Parkin-specific RNAi) provoked a marked reduction in mtDNA content in Drosophila heart tubes, associated with increased RNA abundance of the mitochondrial transcription factor Tfam. Together, these findings suggest that cardiac Parkin deficiency interrupts fly heart tube mitochondrial biogenesis (Bhandari et al. 2014), which is in agreement with the previously postulated role for Parkin as a positive regulator of both mitochondrial biogenesis and mitophagy in Drosophila skeletal muscle (Vincow et al. 2013) and cultured cells (Kuroda et al. 2006; Rothfuss et al. 2009).
In mammalian hearts, the evidence for functional cooperation between mitochondrial biogenesis and Parkin signaling is largely indirect and derived from genetic perturbation of mitochondrial dynamics factors. Compared with the cultured cells in which the fundamentals of mitochondrial dynamism were originally described (Koshiba et al. 2004), adult cardiac myocytes are globally impaired in mitochondrial dynamics; fusion and fission occur so infrequently as to not be observable using normal live-cell techniques, and subcellular trafficking has been inferred from biochemical and functional distinctiveness observed in interfibrillar and subsarcolemmal cardiac mitochondria (Hollander et al. 2014; Song and Dorn 2015). Nevertheless, hearts with cardiomyocyte-specific defects in outer mitochondrial membrane fusion (combined mitofusin 1 [Mfn1] and Mfn2 ablation) or fission (Drp1 ablation) exhibit dysfunction of both mitochondrial biogenesis signaling and mitophagy. In a side-by-side comparative study, cardiac-specific abrogation of either mitochondrial fusion or fission in adult mouse hearts provoked lethal cardiomyopathies over ∼6 wk, but the underlying phenotypes were quite different (Song et al. 2015b). As expected, interrupting mitochondrial fission produced elongated mitochondria, whereas interrupting mitochondrial fusion produced abnormally small, fragmented mitochondria. Mitochondrial biogenesis (Tfam, PGC-1α, and PGC1-β gene expression) was similarly impaired in both fusion- and fission-deficient hearts, but mitophagy was dysregulated in opposite directions; mitophagy (measured as an increase in the autophagy proteins p62/SQSTM1 and LC3 in mitochondria-enriched myocardial protein fractions) was greater in fission-defective Drp1-null hearts but not in fusion-defective Mfn1/Mfn2-double-null hearts (Song et al. 2015b). A central role for Parkin in the mitophagy induced by conditionally interrupting mitochondrial fission (cardiac Drp1 knockout) was subsequently demonstrated through concomitant cardiomyocyte-specific deletion of Parkin and Drp1, which prevented the characteristic hypermitophagy and delayed the dilated cardiomyopathy typically induced by Drp1 ablation (Song et al. 2015a). Together, these findings point to a level of interdependence between mitochondrial biogenesis and Parkin-mediated mitophagy that was not evident from previous investigations.
A plausible mechanism linking mitophagy to mitochondrial biogenesis involves Parkin-interacting substrate (PARIS), which is chronically removed by PINK1-independent Parkin-mediated ubiquitination and therefore accumulates under conditions of Parkin insufficiency (Shin et al. 2011). PARIS regulates mitochondrial biogenesis by transcriptionally repressing PGC-1α, likely through binding of the insulin response sequence (IRS) of the PGC-1α promoter region (Shin et al. 2011). PARIS suppression ameliorated the Parkinson's disease-like pathology provoked by stereotaxic injection of adenoviral-Cre into the nigrostriatal area of Parkin floxed allele mouse brains. Thus, accumulation of PARIS after abrogation of its normal Parkin-mediated removal mechanism caused degeneration of dopaminergic neurons. Furthermore, transgenic overexpression of PARIS is sufficient to induce dopaminergic neuron degeneration, and PGC-1α overexpression can rescue this (Shin et al. 2011). Together, these findings mechanistically link a mitophagy-independent action of Parkin on its substrate, PARIS, to modulate PGC-1α-directed mitochondrial biogenesis (Fig. 3). Moreover, the presence of NRF-1- and NRF-2-occupied promoter elements within the genes encoding mitophagy factors Parkin and PINK1 suggests the existence of additional functional interactions between mitochondrial biogenic regulators and mitophagy (Satoh et al. 2013).
A paradox is posed by the observed interruption of mitophagy after combined cardiac Mfn1 and Mfn2 ablation, in which mitochondrial dysfunction and depolarization that normally stimulate mitophagic signaling are both increased (Chen et al. 2011; Song et al. 2015b). It is likely that interruption of mitophagy is not a function of decreased mitochondrial fusion but is the consequence of a lack of Mfn2 that, after its phosphorylation by PINK1, can function as a mitochondrial binding partner (i.e., receptor) for Parkin (Fig. 3; Chen and Dorn 2013). The seminal observations suggesting a role for Mfn2–Parkin interactions in mitophagy were that Parkin does not localize to depolarized mitochondria in cardiomyocytes of cardiac-specific Mfn2-null mice (Chen and Dorn 2013) or neurons in neuron-specific Mfn2-null mice (Lee et al. 2012a). Subsequent mutation studies revealed that PINK1-mediated phosphorylation of two specific amino acids, Mfn2 T111 and S442, was essential for the Parkin–Mfn2-binding interaction (Chen and Dorn 2013). However, PINK1 phosphorylated Mfn2 cannot be the only mitochondrial Parkin receptor, as Parkin will translocate to depolarized mitochondria of murine embryonic fibroblasts derived from mice with germline Mfn2 deficiency. The identities of other Parkin receptors and the relative role of the PINK1–Mfn2–Parkin signaling axis compared with possible alternative Parkin-dependent and Parkin-independent mitophagy pathways in hearts and other organs remain to be determined.
The findings described above indicate that mitochondrial dynamics affect biogenesis. There is also evidence that the biogenesis circuitry exerts regulation upon dynamics via effects on fusion and fission. As described above, targeted disruption of PGC-1β genes immediately after birth in a generalized PGC-1α-deficient background results in a progressive and lethal cardiomyopathy with severely altered mitochondrial morphology, including fragmentation and elongation, suggestive of defects in proper mitochondrial dynamics (Martin et al. 2014). The levels of several proteins critical for mitochondrial fusion and fission were down-regulated in the postnatal PGC-1α/β-deficient mice, including Mfn1, Mfn2, and Opa1 (Martin et al. 2014). Mfn1 and Mfn2 have both been shown to be direct ERRα/PGC-1α gene targets (Soriano et al. 2006; Martin et al. 2014). These results demonstrate one mechanism for the coordination of mitochondrial biogenesis, dynamics, and maturation in postnatal hearts.
In summary, recent data have implicated cross-regulatory circuits that coordinate mitophagy, mitochondrial dynamics, and mitochondrial biogenesis (Fig. 3). PGC-1α, through coactivation of ERR, PPAR, and other transcription factors, not only controls biogenesis but also regulates components of mitophagy and dynamics. Intriguingly, mitophagy signals also communicate with the biogenic machinery, likely via regulation of PGC-1α by factors such as PARIS. It is possible, if not likely, that other signals exist to coordinately regulate mitophagy, dynamics, and biogenesis. Finally, this circuitry is critical for postnatal maturation of the cardiac mitochondrial system. Akin to the switch from fetal to adult contractile gene expression, it is tempting to speculate that coordination of mitophagy and biogenesis occurs in adult hearts as a quality control mechanism relevant to aging or physiological/pathophysiological stressors. It should be noted that the primary driver of the biogenesis–mitophagy axis during postnatal development remains unknown. Is it triggered by mitophagy, PGC-1-mediated biogenesis, or independent signals? Another important question relates to the nature of relevant upstream signals that initiate this process. Such questions pave the way for important future research endeavors.
Mitochondrial remodeling in disease states
Fuel shifts and energetic remodeling in hypertrophied and failing hearts
Pathological cardiac hypertrophy and heart failure are characterized by an increase in the expression of many contractile and structural genes that are normally expressed in fetal hearts along with decreased expression of normal adult genes. This reactivation of the so-called “fetal gene program” also applies to energy metabolism in heart failure. For instance, expression of PGC-1/PPAR-driven FAO genes is down-regulated during cardiac hypertrophy even prior to overt cardiac dysfunction (Sack et al. 1996; Lai et al. 2014a). Consistent with this, FAO rates and the contribution of β-oxidation-generated acetyl-CoA into the TCA cycle are reduced in pressure overload-induced cardiac hypertrophy and in failing hearts (Akki et al. 2008; Kolwicz et al. 2012; Zhang et al. 2013). In humans, cardiac hypertrophy has also been shown to be associated with reduced myocardial fatty acid utilization (de las Fuentes et al. 2006). These observations indicate that mitochondria are remodeled to a phenotype with reduced capacity for FAO during the development of cardiac hypertrophy and heart failure.
PGC-1/ERR signaling is also decreased in failing hearts in preclinical animal models and humans. Transcriptomic profiling has demonstrated down-regulation of these factors and many of the downstream targets in end-stage failing human heart samples (Gupte et al. 2014). Metabolomic signatures of failing human hearts provide evidence of metabolic dysfunction and a decline in oxidative metabolism with decreased short and medium chain acylcarnitines as well as TCA cycle intermediates (Gupte et al. 2014). Interestingly, similar profiling in earlier stages of heart failure in mice, while demonstrating the expected down-regulation of FAO enzyme gene expression (altered PPAR activity), revealed only minimal evidence for down-regulation of ETC and OXPHOS gene expression (Lai et al. 2014a). Taken together, these results imply progressive, pathologic remodeling and dysregulation of mitochondrial energy production during the development of heart failure. It is likely that deactivation of PPARα and subsequent decreased FAO are early events that contribute to a switch to other fuel substrates, such as glucose and ketones. However, as the disease progresses, expression of ERR and PGC-1 is also down-regulated, resulting in a greater reduction of mitochondrial oxidative capacity. Additionally, the results suggest that post-translational modifications likely contribute to the metabolic derangements during the development of heart failure (Lai et al. 2014a).
Pathologic cardiac remodeling, biogenesis, and mitophagy
Given the known links between mitophagy and mitochondrial biogenesis, what role does mitophagy play during pathologic cardiac remodeling relevant to heart failure? Much less is known about this area, but interest is emerging. As stated above, although unstressed Parkin-deficient mice have normal cardiac function, they accumulate abnormal mitochondria with age (Kubli et al. 2013a). In addition, deletion of Parkin and impaired mitophagy result in sensitization to ischemic injury, as Parkin knockout mice have larger infarcts and pathologic remodeling following myocardial infarction (Kubli et al. 2013a). Parkin levels and mitophagy are also rapidly increased in the border zone following infarct. There is also evidence that increased mitophagy provides a mechanism for protective ischemic preconditioning (Huang et al. 2011). Ischemic preconditioning stimulates Parkin translocation to the mitochondria and is attenuated in Parkin knockout mice (Huang et al. 2011). Given these results, it is tempting to speculate that pathologic remodeling in the heart involves waves of mitochondrial biogenesis and mitophagy. It is also possible that physiological stressors such as exercise or pregnancy could activate rounds of adaptive mitophagy and biogenesis. Answers to these questions may lead to insights into targeting these pathways for therapeutic benefit in the setting of heart failure.
Acknowledgments
We thank Ling Lai for figure preparations and helpful discussion, Teresa Leone for critical reading, and Lorenzo Thomas for assistance in the preparation of this manuscript. This work was supported by National Institutes of Health grants R01 DK045416 and R01 HL058493 to D.P.K., and R01 HL59888 and R01 HL128071 to G.W.D.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.269894.115.
References
- Ahuja P, Zhao P, Angelis E, Ruan H, Korge P, Olson A, Wang Y, Jin ES, Jeffrey FM, Portman M, et al. 2010. Myc controls transcriptional regulation of cardiac metabolism and mitochondrial biogenesis in response to pathological stress in mice. J Clin Invest 120: 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB, Williams RS, Yan Z. 2005. Exercise stimulates Pgc-1α transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem 280: 19587–19593. [DOI] [PubMed] [Google Scholar]
- Akki A, Smith K, Seymour AM. 2008. Compensated cardiac hypertrophy is characterised by a decline in palmitate oxidation. Mol Cell Biochem 311: 215–224. [DOI] [PubMed] [Google Scholar]
- Alaynick WA, Kondo RP, Xie W, He W, Dufour CR, Downes M, Jonker JW, Giles W, Naviaux RK, Giguere V, et al. 2007. ERRγ directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab 6: 13–24. [DOI] [PubMed] [Google Scholar]
- Andersson U, Scarpulla RC. 2001. Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol Cell Biol 21: 3738–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ. 1998. Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor α (PPARα). J Biol Chem 273: 5678–5684. [DOI] [PubMed] [Google Scholar]
- Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. 2006. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-γ coactivator 1α. Proc Natl Acad Sci 103: 10086–10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafian H, Frenneaux MP, Opie LH. 2007. Metabolic mechanisms in heart failure. Circulation 116: 434–448. [DOI] [PubMed] [Google Scholar]
- Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. 2002. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J 16: 1879–1886. [DOI] [PubMed] [Google Scholar]
- Balaban RS. 1990. Regulation of oxidative phosphorylation in the mammalian cell. Am J Physiol 258: C377–C389. [DOI] [PubMed] [Google Scholar]
- Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW II. 2014. Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ Res 114: 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt JM, Djouadi F, Kelly DP. 1998. Fatty acids activate transcription of the muscle carnitine palmitoyltransferase I gene in cardiac myocytes via the peroxisome proliferator-activated receptor α. J Biol Chem 273: 23786–23792. [DOI] [PubMed] [Google Scholar]
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. 2009. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dorn GW II. 2013. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Liu Y, Dorn GW II. 2011. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 109: 1327–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Hazen BC, Russell AP, Kralli A. 2013. Peroxisome proliferator-activated receptor γ coactivator 1 (PGC-1)- and estrogen-related receptor (ERR)-induced regulator in muscle 1 (Perm1) is a tissue-specific regulator of oxidative capacity in skeletal muscle cells. J Biol Chem 288: 25207–25218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. 2006. Drosophila pink1 is required for mitochondrial function and interacts genetically with Parkin. Nature 441: 1162–1166. [DOI] [PubMed] [Google Scholar]
- Coste A, Louet JF, Lagouge M, Lerin C, Antal MC, Meziane H, Schoonjans K, Puigserver P, O'Malley BW, Auwerx J. 2008. The genetic ablation of SRC-3 protects against obesity and improves insulin sensitivity by reducing the acetylation of PGC-1α. Proc Natl Acad Sci 105: 17187–17192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csaki LS, Reue K. 2010. Lipins: multifunctional lipid metabolism proteins. Annu Rev Nutr 30: 257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czubryt MP, McAnally J, Fishman GI, Olson EN. 2003. Regulation of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci 100: 1711–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de las Fuentes L, Soto PF, Cupps BP, Pasque MK, Herrero P, Gropler RJ, Waggoner AD, Davila-Roman VG. 2006. Hypertensive left ventricular hypertrophy is associated with abnormal myocardial fatty acid metabolism and myocardial efficiency. J Nucl Cardiol 13: 369–377. [DOI] [PubMed] [Google Scholar]
- Din S, Konstandin MH, Johnson B, Emathinger J, Volkers M, Toko H, Collins B, Ormachea L, Samse K, Kubli DA, et al. 2014. Metabolic dysfunction consistent with premature aging results from deletion of Pim kinases. Circ Res 115: 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djouadi F, Brandt JM, Weinheimer CJ, Leone TC, Gonzalez FJ, Kelly DP. 1999. The role of the peroxisome proliferator-activated receptor α (PPARα) in the control of cardiac lipid metabolism. Prostaglandins Leukot Essent Fatty Acids 60: 339–343. [DOI] [PubMed] [Google Scholar]
- Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, Evans RM, Blanchette M, Giguere V. 2007. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRα and γ. Cell Metab 5: 345–356. [DOI] [PubMed] [Google Scholar]
- Durcan TM, Fon EA. 2015. The three ‘P's of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev 29: 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, et al. 2002. The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. J Clin Invest 109: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC Jr, Kelly DP. 2006. Lipin 1 is an inducible amplifier of the hepatic PGC-1α/PPARα regulatory pathway. Cell Metab 4: 199–210. [DOI] [PubMed] [Google Scholar]
- Gan Z, Rumsey J, Hazen BC, Lai L, Leone TC, Vega RB, Xie H, Conley KE, Auwerx J, Smith SR, et al. 2013. Nuclear receptor/microRNA circuitry links muscle fiber type to energy metabolism. J Clin Invest 123: 2564–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge K, Guermah M, Yuan CX, Ito M, Wallberg AE, Spiegelman BM, Roeder RG. 2002. Transcription coactivator TRAP220 is required for PPARγ2-stimulated adipogenesis. Nature 417: 563–567. [DOI] [PubMed] [Google Scholar]
- Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. 2007. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J 26: 1913–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleyzer N, Vercauteren K, Scarpulla RC. 2005. Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol Cell Biol 25: 1354–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulick T, Cresci S, Caira T, Moore DD, Kelly DP. 1994. The peroxisome proliferator-activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression. Proc Natl Acad Sci 91: 11012–11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupte AA, Hamilton DJ, Cordero-Reyes AM, Youker KA, Yin Z, Estep JD, Stevens RD, Wenner B, Ilkayeva O, Loebe M, et al. 2014. Mechanical unloading promotes myocardial energy recovery in human heart failure. Circ Cardiovasc Genet 7: 266–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, et al. 2013. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell 23: 302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Sun C, Wang F, Shan A, Guo T, Gu W, Cui B, Ning G. 2012. Peri-implantation lethality in mice lacking the PGC-1-related coactivator protein. Dev Dyn 241: 975–983. [DOI] [PubMed] [Google Scholar]
- Hollander JM, Thapa D, Shepherd DL. 2014. Physiological and structural differences in spatially distinct subpopulations of cardiac mitochondria: influence of cardiac pathologies. Am J Physiol Heart Circ Physiol 307: H1–H14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hom J, Sheu SS. 2009. Morphological dynamics of mitochondria—a special emphasis on cardiac muscle cells. J Mol Cell Cardiol 46: 811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. 2011. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One 6: e20975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo L, Scarpulla RC. 2001. Mitochondrial DNA instability and peri-implantation lethality associated with targeted disruption of nuclear respiratory factor 1 in mice. Mol Cell Biol 21: 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss JM, Kopp RP, Kelly DP. 2002. Peroxisome proliferator-activated receptor coactivator-1α (PGC-1α) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-α and -γ. Identification of novel leucine-rich interaction motif within PGC-1α. J Biol Chem 277: 40265–40274. [DOI] [PubMed] [Google Scholar]
- Huss JM, Torra IP, Staels B, Giguere V, Kelly DP. 2004. Estrogen-related receptor α directs peroxisome proliferator-activated receptor α signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol 24: 9079–9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, Giguere V, Murphy E, Kelly DP. 2007. The nuclear receptor ERRα is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab 6: 25–37. [DOI] [PubMed] [Google Scholar]
- Issemann I, Green S. 1990. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347: 645–650. [DOI] [PubMed] [Google Scholar]
- Jager S, Handschin C, St-Pierre J, Spiegelman BM. 2007. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci 104: 12017–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM, et al. 2014. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 33: 2798–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S. 2014. Integrated physiology and systems biology of PPARα. Mol Metab 3: 354–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee JH, Iyer VR. 2008. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS One 3: e1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolwicz SC Jr, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. 2012. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res 111: 728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. 2004. Structural basis of mitochondrial tethering by mitofusin complexes. Science 305: 858–862. [DOI] [PubMed] [Google Scholar]
- Kressler D, Schreiber SN, Knutti D, Kralli A. 2002. The PGC-1-related protein PERC is a selective coactivator of estrogen receptor α. J Biol Chem 277: 13918–13925. [DOI] [PubMed] [Google Scholar]
- Kubli DA, Quinsay MN, Gustafsson AB. 2013a. Parkin deficiency results in accumulation of abnormal mitochondria in aging myocytes. Commun Integr Biol 6: e24511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. 2013b. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 288: 915–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Cortez MQ, Moyzis AG, Najor RH, Lee Y, Gustafsson AB. 2015. PINK1 is dispensable for mitochondrial recruitment of parkin and activation of mitophagy in cardiac myocytes. PLoS One 10: e0130707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda Y, Mitsui T, Kunishige M, Shono M, Akaike M, Azuma H, Matsumoto T. 2006. Parkin enhances mitochondrial biogenesis in proliferating cells. Hum Mol Genet 15: 883–895. [DOI] [PubMed] [Google Scholar]
- Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP. 2008. Transcriptional coactivators PGC-1α and PGC-1β control overlapping programs required for perinatal maturation of the heart. Genes Dev 22: 1948–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, et al. 2014a. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail 7: 1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai L, Wang M, Martin OJ, Leone TC, Vega RB, Han X, Kelly DP. 2014b. A role for peroxisome proliferator-activated receptor γ coactivator 1 (PGC-1) in the regulation of cardiac mitochondrial phospholipid biosynthesis. J Biol Chem 289: 2250–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau E, Huang D, Cao Q, Dincer TU, Black CM, Lin AJ, Lee JM, Wang D, Liem DA, Lam MP, et al. 2015. Spatial and temporal dynamics of the cardiac mitochondrial proteome. Expert Rev Proteomics 12: 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Sterky FH, Mourier A, Terzioglu M, Cullheim S, Olson L, Larsson NG. 2012a. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum Mol Genet 21: 4827–4835. [DOI] [PubMed] [Google Scholar]
- Lee Y, Dawson VL, Dawson TM. 2012b. Animal models of Parkinson's disease: vertebrate genetics. Cold Spring Harb Perspect Med 2: a009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. 2000. Peroxisome proliferator-activated receptor γ coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest 106: 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone TC, Weinheimer CJ, Kelly DP. 1999. A critical role for the peroxisome proliferator-activated receptor α (PPARα) in the cellular fasting response: the PPARα-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci 96: 7473–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O'Donnell KA, Kim JW, Yustein JT, Lee LA, Dang CV. 2005. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol 25: 6225–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. 2002. Peroxisome proliferator-activated receptor γ coactivator 1β (PGC-1β), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem 277: 1645–1648. [DOI] [PubMed] [Google Scholar]
- Little JP, Safdar A, Cermak N, Tarnopolsky MA, Gibala MJ. 2010. Acute endurance exercise increases the nuclear abundance of PGC-1α in trained human skeletal muscle. Am J Physiol Regul Integr Comp Physiol 298: R912–R917. [DOI] [PubMed] [Google Scholar]
- Lotz C, Lin AJ, Black CM, Zhang J, Lau E, Deng N, Wang Y, Zong NC, Choi JH, Xu T, et al. 2014. Characterization, design, and function of the mitochondrial proteome: from organs to organisms. J Proteome Res 13: 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin OJ, Lai L, Soundarapandian MM, Leone TC, Zorzano A, Keller MP, Attie AD, Muoio DM, Kelly DP. 2014. A role for peroxisome proliferator-activated receptor γ coactivator-1 in the control of mitochondrial dynamics during postnatal cardiac growth. Circ Res 114: 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascaro C, Acosta E, Ortiz JA, Marrero PF, Hegardt FG, Haro D. 1998. Control of human muscle-type carnitine palmitoyltransferase I gene transcription by peroxisome proliferator-activated receptor. J Biol Chem 273: 8560–8563. [DOI] [PubMed] [Google Scholar]
- McMullen PD, Bhattacharya S, Woods CG, Sun B, Yarborough K, Ross SM, Miller ME, McBride MT, LeCluyse EL, Clewell RA, et al. 2014. A map of the PPARα transcription regulatory network for primary human hepatocytes. Chem Biol Interact 209: 14–24. [DOI] [PubMed] [Google Scholar]
- Mitra MS, Schilling JD, Wang X, Jay PY, Huss JM, Su X, Finck BN. 2011. Cardiac lipin 1 expression is regulated by the peroxisome proliferator activated receptor γ coactivator 1α/estrogen related receptor axis. J Mol Cell Cardiol 51: 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. 2008. A mitochondrial protein compendium elucidates complex I disease biology. Cell 134: 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. 2006. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441: 1157–1161. [DOI] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ. 2015. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85: 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosdocimo DA, Anand P, Liao X, Zhu H, Shelkay S, Artero-Calderon P, Zhang L, Kirsh J, Moore D, Rosca MG, et al. 2014. Kruppel-like factor 15 is a critical regulator of cardiac lipid metabolism. J Biol Chem 289: 5914–5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosdocimo DA, John JE, Zhang L, Efraim ES, Zhang R, Liao X, Jain MK. 2015. KLF15 and PPARα cooperate to regulate cardiomyocyte lipid gene expression and oxidation. PPAR Res 2015: 201625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. 1998. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92: 829–839. [DOI] [PubMed] [Google Scholar]
- Ristevski S, O'Leary DA, Thornell AP, Owen MJ, Kola I, Hertzog PJ. 2004. The ETS transcription factor GABPα is essential for early embryogenesis. Mol Cell Biol 24: 5844–5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothfuss O, Fischer H, Hasegawa T, Maisel M, Leitner P, Miesel F, Sharma M, Bornemann A, Berg D, Gasser T, et al. 2009. Parkin protects mitochondrial genome integrity and supports mitochondrial DNA repair. Hum Mol Genet 18: 3832–3850. [DOI] [PubMed] [Google Scholar]
- Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. 1996. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 94: 2837–2842. [DOI] [PubMed] [Google Scholar]
- Satoh J, Kawana N, Yamamoto Y. 2013. Pathway analysis of ChIP-seq-based NRF1 target genes suggests a logical hypothesis of their involvement in the pathogenesis of neurodegenerative diseases. Gene Regul Syst Bio 7: 139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarffe LA, Stevens DA, Dawson VL, Dawson TM. 2014. Parkin and PINK1: much more than mitophagy. Trends Neurosci 37: 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC. 2008. Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann N Y Acad Sci 1147: 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpulla RC, Vega RB, Kelly DP. 2012. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab 23: 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer PJ, Wende AR, Magee CJ, Neilson JR, Leone TC, Chen F, Kelly DP. 2004. Calcineurin and calcium/calmodulin-dependent protein kinase activate distinct metabolic gene regulatory programs in cardiac muscle. J Biol Chem 279: 39593–39603. [DOI] [PubMed] [Google Scholar]
- Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ, Kralli A. 2004. The estrogen-related receptor α (ERRα) functions in PPARγ coactivator 1α (PGC-1α)-induced mitochondrial biogenesis. Proc Natl Acad Sci 101: 6472–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. 2011. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson's disease. Cell 144: 689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirihai OS, Song M, Dorn GW II. 2015. How mitochondrial dynamism orchestrates mitophagy. Circ Res 116: 1835–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoag J, Haq R, Zhang M, Liu L, Rowe GC, Jiang A, Koulisis N, Farrel C, Amos CI, Wei Q, et al. 2013. PGC-1 coactivators regulate MITF and the tanning response. Mol Cell 49: 145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladek R, Bader JA, Giguere V. 1997. The orphan nuclear receptor estrogen-related receptor α is a transcriptional regulator of the human medium-chain acyl coenzyme A dehydrogenase gene. Mol Cell Biol 17: 5400–5409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Dorn GW II. 2015. Mitoconfusion: noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab 21: 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Chen Y, Gong G, Murphy E, Rabinovitch PS, Dorn GW II. 2014. Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res 115: 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Gong G, Burelle Y, Gustafsson AB, Kitsis RN, Matkovich SJ, Dorn GW II. 2015a. Interdependence of Parkin-mediated mitophagy and mitochondrial fission in adult mouse hearts. Circ Res 117: 346–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Mihara K, Chen Y, Scorrano L, Dorn GW II. 2015b. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21: 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano FX, Liesa M, Bach D, Chan DC, Palacin M, Zorzano A. 2006. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-γ coactivator-1α, estrogen-related receptor-α, and mitofusin 2. Diabetes 55: 1783–1791. [DOI] [PubMed] [Google Scholar]
- Stanley WC, Recchia FA, Lopaschuk GD. 2005. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85: 1093–1129. [DOI] [PubMed] [Google Scholar]
- Terada S, Goto M, Kato M, Kawanaka K, Shimokawa T, Tabata I. 2002. Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochem Biophys Res Commun 296: 350–354. [DOI] [PubMed] [Google Scholar]
- van der Meer DL, Degenhardt T, Vaisanen S, de Groot PJ, Heinaniemi M, de Vries SC, Muller M, Carlberg C, Kersten S. 2010. Profiling of promoter occupancy by PPARα in human hepatoma cells via ChIP–chip analysis. Nucleic Acids Res 38: 2839–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, et al. 2013. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 23: 287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega RB, Kelly DP. 1997. A role for estrogen-related receptor α in the control of mitochondrial fatty acid β-oxidation during brown adipocyte differentiation. J Biol Chem 272: 31693–31699. [DOI] [PubMed] [Google Scholar]
- Vega RB, Horton JL, Kelly DP. 2015. Maintaining ancient organelles: mitochondrial biogenesis and maturation. Circ Res 116: 1820–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincow ES, Merrihew G, Thomas RE, Shulman NJ, Beyer RP, MacCoss MJ, Pallanck LJ. 2013. The PINK1–Parkin pathway promotes both mitophagy and selective respiratory chain turnover in vivo. Proc Natl Acad Sci 110: 6400–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, McDonald C, Petrenko NB, Leblanc M, Wang T, Giguere V, Evans RM, Patel VV, Pei L. 2015. Estrogen-related receptor α (ERRα) and ERRγ are essential coordinators of cardiac metabolism and function. Mol Cell Biol 35: 1281–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Fujii H, Takahashi T, Kodama M, Aizawa Y, Ohta Y, Ono T, Hasegawa G, Naito M, Nakajima T, et al. 2000. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor α associated with age-dependent cardiac toxicity. J Biol Chem 275: 22293–22299. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al. 1999. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124. [DOI] [PubMed] [Google Scholar]
- Zhang L, Jaswal JS, Ussher JR, Sankaralingam S, Wagg C, Zaugg M, Lopaschuk GD. 2013. Cardiac insulin-resistance and decreased mitochondrial energy production precede the development of systolic heart failure after pressure-overload hypertrophy. Circ Heart Fail 6: 1039–1048. [DOI] [PubMed] [Google Scholar]