

Abstract
Large (>90%) for gestational age (LGA) fetuses are usually identified incidentally. Detection of the LGA fetus should first prompt the provider to rule out incorrect dates and maternal diabetes. Once this is done, consideration should be given to certain overgrowth syndromes, especially if anomalies are present. The overgrowth syndromes have significant clinical and molecular overlap, and are associated with developmental delay, tumors, and other anomalies. Although genetic causes of overgrowth are considered postnatally, they are infrequently diagnosed prenatally. Here, we review prenatal sonographic findings in fetal overgrowth syndromes, including Pallister-Killian, Beckwith-Wiedemann, Sotos, Perlman, and Simpson-Golabi-Behmel. We also discuss prenatal diagnosis options and recurrence risks.
Keywords: overgrowth, genetic syndrome, prenatal diagnosis, Beckwith-Wiedemann syndrome, Pallister-Killian syndrome, Simpson-Golabi-Behmel syndrome, Sotos syndrome, Weaver syndrome
Introduction
Large (>90%) for gestational age (LGA) fetuses are usually identified during a routine office visit when fundal height is measured, or during an ultrasound examination performed for other reasons. The most common reason for an LGA fetus is maternal diabetes mellitus or incorrect dates. When these two causes are ruled out, attention should be given to other etiologies of fetal overgrowth. Generally, macrosomia is not detectable at the 16 to 20 week anatomy survey. Many fetuses with overgrowth syndromes will fall into the normal weight range until later in gestation. This makes early diagnosis of overgrowth syndromes difficult. However, many fetuses with overgrowth syndromes will have other anomalies detectable in the second trimester. The overgrowth syndromes have significant clinical and molecular overlap, and are associated with developmental delay, tumors, and other anomalies (Baujat et al., 2005). It is our impression that although genetic conditions are considered in the postnatal differential diagnosis of overgrowth, they are relatively infrequently diagnosed prenatally. Genetic causes of overgrowth include chromosomal abnormalities, as well as syndromes such as Sotos, Beckwith-Wiedemann, Perlman, and Simpson-Golabi-Behmel. When an overgrowth syndrome is suspected prenatally, it is essential that a clinical geneticist be involved to assist in the differential diagnosis and selection of appropriate tests. In the majority of these cases, a prenatal diagnosis will not be made. However, increased awareness of the differential diagnosis of overgrowth syndromes can prepare the neonatologist for potential neonatal problems associated with these syndromes. Many overgrowth syndromes exist, but in this review, we will focus on the overgrowth syndromes that have been described with a prenatal presentation and that have clinically available molecular testing.
Considerations in the Differential Diagnosis
Many of the overgrowth syndromes have overlapping clinical and molecular features. A suggested approach to the prenatal diagnosis of overgrowth syndromes is given in Figure 1.
Figure 1.
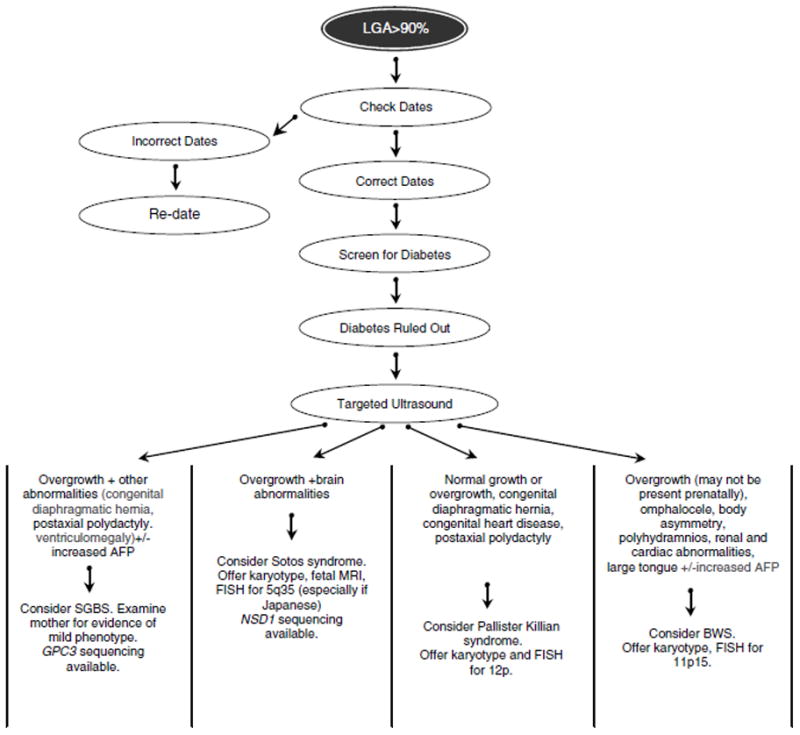
Suggested algorithm for the diagnostic work-up of the fetus with overgrowth and other anomalies.
Chromosomal Abnormalities
Several chromosomal abnormalities are associated with fetal overgrowth, including trisomy 12p, mosaic tetrasomy 12p (also known as Pallister-Killian syndrome), trisomy 4p16.3, trisomy 5p, trisomy 15q25, mosaic trisomy 8 and monosomy 22q13 (Segel et al., 2006, Cohen et al., 2005). With the introduction of chromosome microarrays into clinical medicine, chromosome abnormalities associated with overgrowth will be more commonly found. For example, a case report described two unrelated patients with trigonocephaly, overgrowth, and macrocephaly with a de novo microdeletion in 9q22.32–q22.33 found by array comparative genomic hybridization (Redon et al., 2006). Prenatal microarrays are currently available that can diagnose microdeletions in the following genomic regions; Sotos (5q35), Beckwith Wiedemann (11p15), and 22q13. A prenatal microarray study may be useful if there is evidence of overgrowth with other abnormalities. Clinical trials are in progress to determine the clinical utility of prenatal micorarrays.
Pallister-Killian syndrome
Pallister Killian syndrome (Online Mendelian Inheritance in Man [OMIM] #601803) is caused by mosaicism for tetrasomy of chromosome 12p. Clinical features include severe mental retardation, hypo- or hyperpigmentation, seizures, postaxial polydactyly, and facial dysmorphism, including prominent forehead with sparse anterior scalp hair, hypertelorism, flat occiput, short, anteverted nose, and short neck. Skeletal anomalies, congenital heart defects, and congenital diaphragmatic hernia are also seen in this condition. Pallister-Killian syndrome is associated with advanced maternal age, and is thought to be caused by non-disjunction, followed by an abnormal centromere division in meiosis I or II (Struthers et al., 1999). This results in an isochromosome 12p. Tetrasomy 12p can be detected by amniocentesis and confirmed with FISH using specific probes for 12p. The detection of tetrasomy 12p is affected by the tissue type. It is more readily detected in amniocytes and skin because it is thought that peripheral blood lympocytes lose the extra chromosome due to their more frequent cell division. In an affected infant, if the peripheral blood karyotype is normal, then skin biopsy or karyotype of other tissue sources should be considered. Recurrence risk is low because this is a sporadic condition.
The most common prenatal sonographic findings seen in Pallister-Killian syndrome and other conditions are listed in Table 1 [Doray et al., 2002; Bresson et al., 1991, Paladini et al., 2000; Liberati et al., 2008)]. It is important to distinguish this syndrome, which occurs sporadically, from Fryns syndrome, an autosomal recessive condition, which is also associated with skeletal anomalies and diaphragmatic hernia. Patients with Fryns syndrome, however, typically have intrauterine growth restriction and die more often in the newborn period. Significantly, intrauterine growth restriction has never been reported in Pallister-Killian syndrome.
Table 1.
Differential Diagnosis of Genetic Overgrowth Syndromes
| Syndrome | Gene location | Inheritance | Possible sonographic findings | Other aids to diagnosis |
|---|---|---|---|---|
| Beckwith-Wiedemann | 11p15 | Variable; sporadic in 85% of cases but can be autosomal dominant; also can be caused by uniparental disomy and imprinting defects | Macrosomia, polyhydramnios, omphalocele, enlarged tongue, placentomegaly, long umbilical cord, enlarged echogenic kidneys, pancreatic cystic dysplasia | Karyotype but <1% of BWS have cytogenetically detectable abnormalities; examination of parents by clinical geneticist |
| Pallister-Killian | Tetrasomy 12p | Sporadic | Polyhydramnios, rhizomelic micromelia, congenital heart disease, diaphragmatic hernia (Figure 3), increased nuchal translucency, facial abnormalities | Karyotype; confirm diagnosis with FISH probes for 12p |
| Sotos | NSD1 on 5q35 | Sporadic in 95% of cases | Brain abnormalities, macrocephaly | Fetal MRI; Karyotype, FISH for 5q35, NSD1 sequencing |
| Perlman | Unknown | Autosomal recessive | Polyhydramnios, macrosomia, visceromegaly, enlarged echogenic kidneys, cystic hygroma | |
| Simpson-Golabi-Behmel | GPC3 on Xp26 | X-linked recessive | Polyhydramnios, omphalocele, cystic hygroma, diaphragmatic hernia, enlarged or cystic kidneys, agenesis of the corpus callosum, Dandy Walker malformation, and macrosomia, placentomegaly | Elevated MSAFP level; examination of mother by clinical geneticist; determine fetal gender; GPC3 sequencing |
Beckwith-Wiedemann Syndrome
Beckwith-Wiedemann syndrome (BWS) has an incidence of 1/13,700 and is the most common overgrowth syndrome. Underreporting of minor cases may underestimate its true incidence (Carlin et al., 1990). BWS (OMIM catalog #130650) is characterized by macrosomia, macroglossia, visceromegaly, neonatal hypoglycemia, omphalocele, embryonal tumors, ear creases and pits, renal abnormalities, and adrenocortical cytomegaly (Elliott et al., 1994). Patients with BWS have an increased risk of Wilms’ tumor, hepatoblastoma, neuroblastoma, rhabdomyosarcoma, and adrenocortical carcinoma (Wiedemann, 1983). It is important for affected children to have abdominal sonographic screening for tumors every three months until age eight, after which tumor development is uncommon. Serum alphafetoprotein (AFP) levels should also be checked in the first few years of life so that hepatoblastoma can be detected early.
Approximately twenty cases of prenatally diagnosed BWS have been reported in the literature (Williams et al., 2005). The most commonly described sonographic findings are included in Table 1. (Williams et al., 2005, Cohen, 2005, Le Caignec et al., 2004, Fremond et al., 1997). A targeted ultrasound examination is recommended to evaluate for body asymmetry, abdominal wall defects, renal and cardiac anomalies, macroglossia, adrenomegaly, and an enlarged pancreas (Williams et al., 2005).
An antenatal diagnosis can prepare neonatologists for problems specific to BWS, such as airway compromise due to macroglossia, and severe hypoglycemia (Williams et al., 2005; Pettenati et al., 1986). Many neonatal intensive care units regularly check blood sugars on LGA infants because of the risk of undiagnosed maternal gestational diabetes. This kind of protocol will also prevent complications in neonates with BWS or other genetic syndromes. The reported perinatal mortality rate in BWS from prematurity, macroglossia, and cardiomyopathy is as high as 20% (Williams et al., 2005). Also, the diagnosis may affect mode of delivery, depending on the degree of macrosomia.
The genetics of BWS are quite complex and the diagnosis relies primarily on clinical findings. This complexity can cause difficulty in obtaining a molecular diagnosis. Ten to twenty percent of patients with BWS have paternal uniparental disomy (UPD) (Cyntrynbaum et al., 2005). The majority of these patients have segmental paternal UPD for 11p15, suggesting that there was a post-zygotic recombination that caused the mosaicism. UPD may be difficult to detect because of low level mosaicism. Examination of other tissues, such as skin, may be needed.
BWS is also caused by changes in genes involved in growth regulation on chromosome 11p15, which are also susceptible to imprinting. Imprinting or methylation defects affecting several different genes have also been reported to cause BWS. The imprinting defects in BWS have been well described by other authors and will not be discussed in detail here. (Cooper et al., 2005; Bliek et al., 2001; Weksberg et al., 2001; Cytrynbaum et al., 2005; Debaun et al., 2003; Lam et al., 1999).
There is obvious difficulty in making an antenatal diagnosis of uniparental disomy (UPD) or imprinting defects. However, if macrosomia and other features of BWS are noted prenatally, FISH analysis and karyotype can be performed to detect translocations or inversions that affect genes in the 11p15 region (Reish et al., 2002). Less than 1–2% of cases of BWS have cytogenetically detectable abnormalities in 11p15 (Slavotinek et al., 1997). Translocations and inversions are usually maternally inherited but can arise de novo; thus, parental samples should always be submitted with prenatal samples because of the possibility of familial chromosomal rearrangements, deletions, or duplications. The fastest and most accurate method of detecting BWS is methlyation specific multiplex ligation dependent probe amplication (MS-MLPA) but use of this method prenatally is limited and has not been well described prenatally (Priolo et al., 2008).
Although most cases of BWS are sporadic (up to 85%), 10–15% have autosomal dominant inheritance with preferential maternal transmission (Cohen 2005). Because there is variable penetrance, a thorough family history and physical examination of both parents for hemihypertrophy, earlobe grooves and posterior helical pits is recommended. Pregnant women carrying fetuses with sonographic findings suspicious for BWS should be counseled about all aspects of BWS, including the risk of embryonal tumors. With uniparental disomy the recurrence risk is very low (<1%), because the UPD is thought to arise from a post-zygotic somatic recombination. If no genetic etiology is identified, the risk to future offspring is low, but there should be a discussion with the parents about the possibility of gonadal mosaicism and the limitations of currently available testing.
Sotos Syndrome
Sotos syndrome (OMIM #117550), also known as cerebral gigantism, was first described in 1964 (Sotos et al., 1964). Sotos syndrome is characterized by pre and postnatal overgrowth, developmental delay, and a typical facial appearance that includes macrocephaly, dolichocephaly, a prominent forehead, large hands and feet, advanced bone age, prominent jaw, and variable psychomotor developmental delay (Sotos et al., 1964). Most cases of Sotos syndrome are sporadic, although familial cases have been reported. When inherited, it is thought to be an autosomal dominant disorder with variable penetrance.
Case reports of prenatal findings of Sotos syndrome have been published (Chen et al., 2002; Lemire et al., 2008), and are summarized in Table 1. Sotos syndrome is associated with certain brain abnormalities. One author found that all patients with Sotos syndrome had abnormal MRI scans (Gusmao Melo et al., 2000). The postnatal neuroimaging abnormalities include enlargement of the lateral ventricles, the trigones, and the occipital horns, midline defects, including abnormalities of the corpus callosum with complete or partial agenesis of hypoplasia, as well as other abnormalities (Schaefer et al., 1997; Gusmao Melo et al., 2000). However, brain abnormalities in Sotos syndrome are nonspecific (Tatton-Brown et al., 2005) and many fetuses with Sotos syndrome will not be large for gestational age at the time of level II sonographic examination (16–20 weeks), making it very difficult to diagnose this condition prenatally. If brain abnormalities with or without overgrowth are noted by prenatal sonography, fetal MRI should be obtained.
Sotos syndrome is caused by haploinsufficiency of the NSD1 (nuclear receptor SET domain containing gene 1) gene on chromosome 5q35. To date, more than 100 mutations have been identified in NSD1. Most of the mutations have a truncating effect on the protein (Faravelli et al., 2005).
Many reports have indicated that up to 80 to 90% of patients with Sotos syndrome have an intragenic mutation or a submicroscopic microdeletion that impairs the function of NSD1 (Kurotaki et al., 2002; Douglas et al., 2003;, Kamimura et al., 2003;, Rio et al., 2003; Visser et al., 2003). Routine karyotype analysis usually shows normal results. Non-Japanese patients have an intragenic mutation in 27–93% of cases. The variability in detection rates is the result of the level of expertise of the clinicians who referred the patients for testing. In contrast, only 12% of Japanese patients have intragenic mutations in the NSD1 gene. Fifty percent of Japanese patients have microdeletions in 5q35 that can be detected by FISH, whereas only 10% of non-Japanese patients have this microdeletion (Kurotaki et al., 2003; Tatton-Brown et al., 2005). It has been noted that the microdeletions occur more often in the paternally inherited chromosome (Miyake et al., 2003; Faravelli et al., 2005).
In summary, when Sotos syndrome is suspected prenatally, a metaphase karyotype should be performed to determine whether there is a translocation. NSD1 sequencing and FISH for 5q35 microdeletion are available clinically. If there is an affected member of the family, a mutation in a proband should be identified before prenatal diagnosis is performed. However, more than 95% of cases of Sotos syndrome are secondary to a de novo mutation, so this is only possible in a very small percentage of cases. If neither parent has Sotos syndrome, the risk to future offspring is low (<1%). However, the patient should receive counseling regarding the possibility of germline mosaicism. Offspring of an affected parent have a 50% risk of Sotos syndrome.
Perlman Syndrome
Perlman syndrome is a rare autosomal recessive fetal overgrowth syndrome associated with a high fetal and neonatal mortality rate (64%). Survivors have a high incidence of Wilms’ tumor and mental retardation (Perlman et al., 1973). Perlman syndrome is characterized by dysmorphic facial features, including small mouth, and small upturned nose with a deep crease over the nasal bridge. Other features include bilateral renal enlargement with fetal macrosomia, renal hamartomas, hydronephrosis, hydroureter, developmental delay, and cryptorchidism.
There have been a few reports of abnormal prenatal sonographic findings in Perlman syndrome (Table 1) (Chitty et al., 1998; DeRoche et al., 2004, van der Stege et al., 1998; Henneveld et al., 1999). Significant overlap between Perlman, Simpson-Golabi-Behmel, and Beckwith Wiedemann syndromes exists. All three should be considered in the differential diagnosis of overgrowth with visceromegaly. Distinction between the three can be made postnatally by a medical geneticist.
Parents should be counseled about the 25% recurrence risk if a postnatal diagnosis of Perlman syndrome is made. The etiology of Perlman syndrome is unknown at present and no diagnostic testing is available.
Simpson-Golabi-Behmel Syndrome
Simpson-Golabi-Behmel syndrome (SGBS) is an X-linked recessive condition associated with prenatal and postnatal overgrowth, macrocephaly, congenital diaphragmatic hernia, coarse facies, supernumerary nipples, palatal abnormalities, congenital heart defects, and generalized hypotonia (Xuan et al., 1999). There are also variable visceral, skeletal, and neurological abnormalities present, including ventriculomegaly. Hypoglycemia can manifest in the newborn period. Although normal intelligence has been described, mild to severe mental retardation is common. There is variable expressivity, ranging from mild forms in carrier females to infantile lethal forms. As many as 50% of affected males die in the neonatal period (Neri et al., 1998). Because these patients are at increased risk of tumor development in early childhood, they should have regular sonographic surveillance (Neri et al., 1998). Five types of tumors have been described: Wilms tumor, hepatoblastoma, adrenal neuroblastoma, gonadoblastoma, and hepatocellular carcinoma. There is significant clinical overlap with other overgrowth syndromes, especially BWS and Perlman syndrome.
Only a few case reports of prenatal findings in SGBS are available (Table 1) (Hughes-Benzie et al., 1994; Yamashita et al., 1995). There have been three reports of elevated MSAFP levels in SGBS (none of which had an abdominal wall or spine defect) (Chen et al., 1993, Hughes-Benzie et al., 1994). Hughes-Benzie et al. presented a case report of SGBS, and suggested that fetal macrosomia with a low head to abdominal circumference ratio (4 SD below the mean) and elevated MSAFP level may be antenatal markers for SGBS.
SGBS is an X linked recessive disorder with variable expression in males caused by mutations in the GPC3 gene on Xp26. DNA analysis is available on a clinical basis. The sensitivity of detection of an abnormality ranges from 37% (Li et al., 2001) to 70% (Lin et al., 1999; Veugelers et al., 2000). All males with a mutation in GPC 3 will have clinical findings of SGBS. Carrier females may have certain manifestations of SGBS including accessory nipples, narrow palpebral fissures, extra lumbar and thoracic vertebrae, prominent chin, hypoplastic fingernails, upturned nose, coccygeal skin tags and bony appendages (Golabi et al., 1984). If the mother of an affected child, has a mutation, the chance of transmitting the disease causing mutation is 50%. Thus, 50% of male children will be affected and 50% of female children will be carriers. Prenatal testing is also available if there is a known mutation in the family.
In the setting of a positive family history, prenatal testing should first determine the fetal sex by noninvasive testing using cell free fetal DNA to determine whether markers for the Y chromosome are present in maternal blood. CVS or amniocentesis can also be performed to determine fetal sex if noninvasive prenatal diagnosis is not clinically available. If the karyotype is 46, XY, then DNA from fetal cells can be analyzed for the known familial mutation. Preimplantation genetic diagnosis (PGD) is also available in families that have been molecularly characterized.
Other Syndromes
Costello syndrome (OMIM #218040) is an autosomal dominant condition associated with multiple congenital anomalies, mental retardation, and prenatal overgrowth. There is a characteristic facial appearance that includes macrocephaly, full lips, large mouth, epicanthal folds, and full cheeks. The face becomes “coarse” over time postnatally. Other typical features include deep palmar and plantar creases, nasal papillomata, and loose skin over the back of the hands. There is beginning to be an appreciation of the fetal phenotype in Costello syndrome (Lin et al., 2009; Smith et al., 2009), which includes overgrowth, polyhydramnios, macrocephaly, large for gestational age, shortened long bones, ventriculomegaly, and cardiac arrhythmias. If the above abnormalities are seen antenatally, prenatal DNA diagnosis for HRAS mutations should be considered. Missense mutations in exon 2 of the HRAS gene are found in 80–90% of affected individuals. If no mutation is found in exon 2, then full sequencing of the gene should be performed.
Weaver syndrome (OMIM #277590) is characterized by prenatal overgrowth, developmental delay, macrocephaly, camptodactyly, distinctive facial features, broad thumbs, loose skin, a small chin with a deep skin crease, metaphyseal widening, and advanced bone age (Weaver et al., 1974). There is phenotypic overlap between Sotos syndrome and Weaver syndrome, especially during infancy, but experienced dysmorphologists can appreciate the differences. Some authors have suggested that Sotos syndrome and Weaver syndrome are allelic (Opitz et al., 1998).
Although Weaver syndrome and Sotos syndrome are phenotypically similar, the brain abnormalities noted in Sotos syndrome are specific to Sotos syndrome. To date, there are no reports of prenatal findings in Weaver syndrome, but macrocephaly and overgrowth would also be expected.
NSD1 mutations have been demonstrated in patients with the diagnosis of Weaver syndrome. Over time, the phenotype of the Weaver syndrome patients with the NSD1 mutations strongly resembles the phenotype of Sotos syndrome. However, the patients considered to have “classic” Weaver syndrome did not have mutations in the NSD1 gene, suggesting that a separate gene may be involved in this condition (Douglas et al., 2003; Tatton-Brown et al., 2005).
Another overgrowth syndrome called Macrocephaly Cutis Marmorata Telangiectasia Congenita (M-CMTC) is associated with macrocephaly, neurologic abnormalities (Chiari malformations, ventriculomegaly, hemimegalencephaly), asymmetry, and polydactyly/syndactyly (Moore et al., 1997). This should be considered in the differential diagnosis when the above abnormalities are noted.
Summary and Recommendations
If the fetus is noted to be LGA, dating of the pregnancy should be confirmed, and maternal diabetes mellitus should be excluded. Once these two issues are ruled out, consideration should then be given to other etiologies. Constitutional overgrowth is possible, especially in obese mothers. However, genetic overgrowth syndromes should be included in the differential diagnosis, especially when other abnormalities are noted on sonography or serum screen.
Because these syndromes are rare, we recommend that a medical geneticist be involved in a suspected case of overgrowth to assist in selection and interpretation of molecular testing. Even if a definitive diagnosis is not made prenatally, the neonatal team will be prepared to deal with problem such as hypoglycemia if the obstetrician communicates to the neonatologist that an overgrowth syndrome is suspected. Also, it should be noted that the turn around time for many of the molecular tests may be several weeks. Thus, the definitive diagnosis may not be available until after birth.
We have developed an algorithm for providers who identify overgrowth and other anomalies on prenatal ultrasound examination. An increased awareness of these syndromes among obstetricians will improve their prenatal detection rates and allow pregnant patients to make more informed choices.
Figure 2.
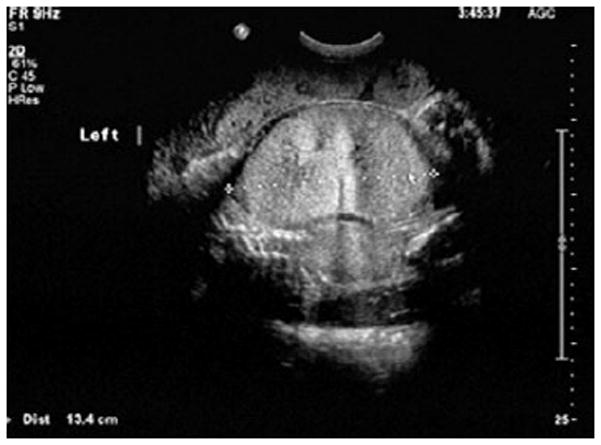
Prenatal sonographic image from a fetus at 32 weeks’ gestation showing renal enlargement and hyperechogenicity. This kidney measures 13 cm in the transverse view. The normal value at this gestational age is 3.8 cm.
Figure 3.
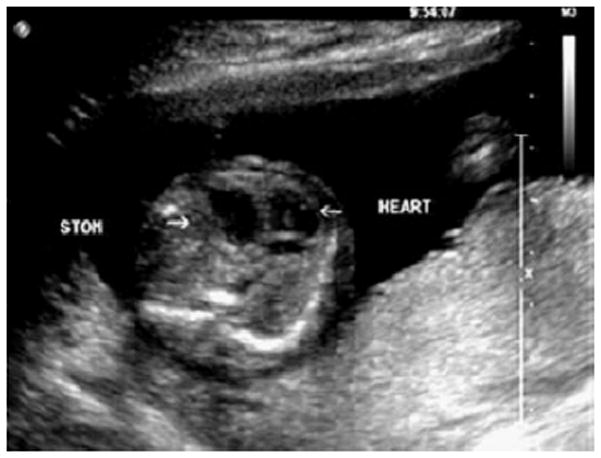
Prenatal sonographic image from a fetus at eighteen weeks’ gestation with congenital diaphragmatic hernia (CDH). When CDH and overgrowth are seen in combination, a karyotype should be performed to look for Pallister-Killian syndrome. SGBS should also be considered.
Acknowledgments
This work was supported by NIH grant T32 HD049341 to Dr. Bianchi.
References
- Baujat G, Rio M, Roosignol S, et al. Clinical and molecular overlap in overgrowth syndromes. Am J Med Genet C Semin Med Genet. 2005;137 :4–11. doi: 10.1002/ajmg.c.30060. [DOI] [PubMed] [Google Scholar]
- Bliek J, Maas SM, Ruijter JM, et al. Increased tumour risk for BWS patients correlates with aberrant H19 and not KCNQ1OT1 methylation: occurrence of KCNQ1OT1 hypomethylation in familial cases of BWS. Hum Mol Genet. 2001;10 :467–76. doi: 10.1093/hmg/10.5.467. [DOI] [PubMed] [Google Scholar]
- Bresson JL, Arbez-Gindre F, Peltie J, et al. Pallister Killian mosaic tetrasomy 12p syndrome: another prenatally diagnosed case. Prenat Diagn. 1991;11 :271–275. doi: 10.1002/pd.1970110409. [DOI] [PubMed] [Google Scholar]
- Carlin ME, Escobar LF, Ward RE, et al. A reassessment of Beckwith-Wiedemann syndrome. Am J Hum Genet. 1990;47(Suppl):A50. [Google Scholar]
- Chen CP, Lin SP, Chang TY, et al. Perinatal imaging findings of inherited Sotos syndrome. Prenat Diagn. 2002;22:887–892. doi: 10.1002/pd.433. [DOI] [PubMed] [Google Scholar]
- Chen E, Johnson JP, Cox VA, et al. Simpson-Golabi-Behmel syndrome: congenital diaphragmatic hernia and radiologic findings in two patients and follow-up of a previously reported case. Am J Med Genet. 1993;46 :574–8. doi: 10.1002/ajmg.1320460523. [DOI] [PubMed] [Google Scholar]
- Chitty LS, Clark T, Maxwell D. Perlman syndrome—a cause of enlarged, hyperechogenic kidneys. Prenat Diagn. 1998;18(11 ):1163–8. doi: 10.1002/(sici)1097-0223(199811)18:11<1163::aid-pd408>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Cohen MM., Jr Beckwith-Wiedemann syndrome: historical, clinicopathological, and etiopathogenetic perspectives. Pediatric and Developmental Pathology. 2005;8 :287–304. doi: 10.1007/s10024-005-1154-9. [DOI] [PubMed] [Google Scholar]
- Cohen MM, Jr, Neri G, Weksberg R. Overgrowth Syndromes. Oxford: Oxford University Press; 2005. Chromosomal Disorders with Overgrowth; pp. 161–165. [Google Scholar]
- Cooper WN, Luharia A, Evans GA, et al. Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2005;13 :1025–1032. doi: 10.1038/sj.ejhg.5201463. [DOI] [PubMed] [Google Scholar]
- Cytrynbaum CS, Smith AC, Rubin T, et al. Advances in overgrowth syndromes: clinical classification to molecular delineation in Sotos syndrome and Beckwith-Wiedemann syndrome. Curr Opin Pediatr. 2005;17 :740–746. doi: 10.1097/01.mop.0000187191.74295.97. [DOI] [PubMed] [Google Scholar]
- Debaun MR, Niemitz EL, Feinberg AP. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet. 2003;72 :156–160. doi: 10.1086/346031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRoche ME, Craffey A, Greenstein R, et al. Antenatal sonographic features of Perlman syndrome. J Ultrasound Med. 2004;23(4):561–4. doi: 10.7863/jum.2004.23.4.561. [DOI] [PubMed] [Google Scholar]
- Doray B, Girard-Lemaire F, Gasser B, et al. Pallister-Killian syndrome: difficulties of prenatal diagnosis. Prenat Diagn. 2002;22 :470–7. doi: 10.1002/pd.342. [DOI] [PubMed] [Google Scholar]
- Douglas J, Hanks S, Temple IK, et al. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet. 2003;72 :132–143. doi: 10.1086/345647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott M, Bayly R, Cole T, et al. Clinical features and natural history of Beckwith-Wiedemann syndrome: presentation of 74 new cases. Clin Genet. 1994;46 :168–174. doi: 10.1111/j.1399-0004.1994.tb04219.x. [DOI] [PubMed] [Google Scholar]
- Faravelli F. NSD1 mutations in Sotos syndrome. Am J Med Gen Part C (Semin Med Genet) 2005;137C :24–31. doi: 10.1002/ajmg.c.30061. [DOI] [PubMed] [Google Scholar]
- Fremond B, Poulain P, Odent S, et al. Prenatal detection of a congenital pancreatic cyst and Beckwith-Wiedemann syndrome. Prenat Diagn. 1997;17 :276–80. [PubMed] [Google Scholar]
- Golabi M, Rosen L. A new X-linked mental retardation-overgrowth syndrome. Am J Med Genet. 1984;17:345–358. doi: 10.1002/ajmg.1320170128. [DOI] [PubMed] [Google Scholar]
- Gusmao Melo D, Pina-Neto JM, Acosta AX, et al. Neuroimaging and echocardiographic findings in Sotos syndrome. Am J Med Genet. 2000;90 :432–433. [PubMed] [Google Scholar]
- Henneveld HT, van Lingen RA, Hamel BC, et al. Perlman syndrome: four additional cases and review. Am J Med Genet. 1999;86(5 ):439–46. [PubMed] [Google Scholar]
- Hughes-Benzie RM, Tolmie JL, Mcnay M, et al. Simpson-Golabi-Behmel syndrome: Disproportionate fetal overgrowth and elevated maternal serum alpha-fetoprotein. Prenat Diagn. 1994;14 :313–318. doi: 10.1002/pd.1970140414. [DOI] [PubMed] [Google Scholar]
- Kamimura J, Endo Y, Kurotaki N, et al. Online mutation report: Identification of 8 novel NSD1 mutations in Sotos syndrome. J Med Genet. 2003;40 :e126. doi: 10.1136/jmg.40.11.e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurotaki N, Imaizumi K, Harada N, et al. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet. 2002;30 :365–366. doi: 10.1038/ng863. [DOI] [PubMed] [Google Scholar]
- Kurotaki N, Harada N, Shimokawa O, et al. Fifty microdeletions among 112 cases of Sotos syndrome: low copy repeats possibly mediate the common deletion. Hum Mutat. 2003;22:378–87. doi: 10.1002/humu.10270. [DOI] [PubMed] [Google Scholar]
- Lam WW, Hatada I, Ohishi S, et al. Analysis of germ line CDKN1C(p57KIP2) mutations in familial and sporadic Beckwith-Wiedemann syndrome (BWS) provides a novel genotype-phenotype correlation. J Med Genet. 1999;36 :518–23. [PMC free article] [PubMed] [Google Scholar]
- Le Caignec C, Gicquel C, Gubler MC, et al. Sonographic findings in Beckwith-Wiedemann syndrome related to H19 hypermethylation. Prenat Diagn. 2004;24 :165–168. doi: 10.1002/pd.818. [DOI] [PubMed] [Google Scholar]
- Lemire EG, Thomas A. Sotos syndrome: Antenatal presentation. Am J Med Genet Part A. 2008;146A :1312–1313. doi: 10.1002/ajmg.a.32283. [DOI] [PubMed] [Google Scholar]
- Li M, Shuman C, Fei YL, et al. GPC3 mutation analysis in a spectrum of patients with overgrowth expands the phenotype of Simpson-Golabi-Behmel syndrome. Am J Med Genet. 2001;102 :161–8. doi: 10.1002/1096-8628(20010801)102:2<161::aid-ajmg1453>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Liberati M, Melchiorre K, D’Emilio I, et al. Fetal facial profile in Pallister-Killian syndrome. Fetal Diagn Ther. 2008;23 :15–7. doi: 10.1159/000109220. [DOI] [PubMed] [Google Scholar]
- Lin AE, Neri G, Hughes-Benzie R, et al. Cardiac anomalies in the Simpson-Golabi-Behmel syndrome. Am J Med Genet. 1999;83 :378–81. [PubMed] [Google Scholar]
- Lin A, O’Brien B, Demmer L, et al. Prenatal features of Costello syndrome: Ultrasonographic findings and atrial tachycardia. Prenat Diagn. 2009 doi: 10.1002/pd2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake N, Kurotaki N, Sugawara H, et al. Preferential paternal origin of microdeletions caused by prezygotic chromosome or chromatid rearrangements in Sotos syndrome. Am J Hum Genet. 2003;72 :1331–1337. doi: 10.1086/375166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CA, Toriello HA, Abuelo DN, et al. Macrocephaly-cutis marmorata telangiectatica congenita: a distinct disorder with developmental delay and connective tissue abnormality. Am J Med Genet. 1997;70 :67–73. [PubMed] [Google Scholar]
- Neri G, Gurrieri F, Zanni G, et al. Clinical and Molecular Aspects of the Simpson-Golabi-Behmel syndrome. Am J Med Genet. 1998;79 :279–283. doi: 10.1002/(sici)1096-8628(19981002)79:4<279::aid-ajmg9>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Opitz JM, Weaver DW, Reynolds JF., Jr The syndromes of Sotos and Weaver: Reports and review. Am J Med Genet. 1998;79 :294–304. doi: 10.1002/(sici)1096-8628(19981002)79:4<294::aid-ajmg12>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Paladini D, Borghese A, Arienzo M, et al. Prospective ultrasound diagnosis of Pallister Killian syndrome in the second trimester of pregnancy: the importance of fetal facial profile. Prenat Diagn. 2000;20 :996–998. [PubMed] [Google Scholar]
- Perlman M, Goldberg GM, Bar-Ziv J, et al. Renal hamartomas and nephroblastomatosis with fetal gigantism: a familial syndrome. J Pediat. 83:414–418. doi: 10.1016/s0022-3476(73)80264-1. [DOI] [PubMed] [Google Scholar]
- Pettenati MJ, Haines JL, Higgins RR, et al. Wiedemann-Beckwith syndrome: presentation of clinical and cytogenetic data on 22 new cases and review of the literature. Hum Genet. 1986;74 :143–154. doi: 10.1007/BF00282078. [DOI] [PubMed] [Google Scholar]
- Priolo M, Sparago A, Mammi C, et al. MS-MLPA is a specific and sensitive technique for detecting all chromosome 11p15.5 imprinting defects of BWS and SRS in a single-tube experiment. Eur J Hum Genet. 2008;16:565–571. doi: 10.1038/sj.ejhg.5202001. [DOI] [PubMed] [Google Scholar]
- Redon R, Baujat G, Sanlaville D, et al. Interstitial 9q22.3 microdeletion: clinical and molecular characterisation of a newly recognised overgrowth syndrome. Eur Journ Hum Genet. 2006;14 :759–767. doi: 10.1038/sj.ejhg.5201613. [DOI] [PubMed] [Google Scholar]
- Reish O, Lerer I, Amiel A, et al. Wiedemann-Beckwith syndrome: further prenatal characterization of the condition. Am J Med Genet. 2002;107 :209–213. doi: 10.1002/ajmg.10143. [DOI] [PubMed] [Google Scholar]
- Rio M, Clech L, Amiel J, et al. Spectrum of NSD1 mutations in Sotos and Weaver syndromes. J Med Genet. 2003;49 :438–440. doi: 10.1136/jmg.40.6.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer GB, Bodensteiner JB, Buehler BA, et al. Neuroimaging findings in Sotos syndrome. Am J Med Genet. 1997;68 :462–465. doi: 10.1002/(sici)1096-8628(19970211)68:4<462::aid-ajmg18>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Segel R, Peter I, Demmer L, et al. The natural history of trisomy 12p. Am J Med Genet. 2006;140A :695–703. doi: 10.1002/ajmg.a.31143. [DOI] [PubMed] [Google Scholar]
- Slavotinek A, Gaunt L, Donnai D. Paternally inherited duplications of 11p15.5 and Beckwith-Wiedemann syndrome. J Med Genet. 1997;34 :819–26. doi: 10.1136/jmg.34.10.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LP, Podraza J, Proud VK. Polyhydramnios, fetal overgrowth, and macrocephaly: prenatal ultrasound findings of Costello syndrome. Am J Med Genet. 2009;149A:779–784. doi: 10.1002/ajmg.a.32778. [DOI] [PubMed] [Google Scholar]
- Sotos JF, Dodge PR, Muirhead D, et al. Cerebral gigantism in childhood. A syndrome of excessively rapid growth and acromegalic features and a nonprogressive neurologic disorder. N Engl J Med. 1964;271 :109–116. doi: 10.1056/NEJM196407162710301. [DOI] [PubMed] [Google Scholar]
- Struthers JL, Cuthbert CD, Khalifa MM. Parental origion of the isochromosome 12p in Pallister-Killian syndrome: molecular analysis of one patient and review of the reported cases. Am J Med Genet. 1999;84 :111–115. [PubMed] [Google Scholar]
- Tatton-Brown K, Douglas J, Coleman K, et al. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. Am J Hum Genet. 2005;77 :193–204. doi: 10.1086/432082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Stege JG, van Eyck J, Arabin B. Prenatal ultrasound observations in subsequent pregnancies with Perlman syndrome. Ultrasound Obstet Gynecol. 1998;11(2 ):149–151. doi: 10.1046/j.1469-0705.1998.11020149.x. [DOI] [PubMed] [Google Scholar]
- Veugelers M, Vermeesch J, Watanabe K, et al. GPC4, the gene for human K-glypican, flanks GPC3 on xq26: deletion of the GPC3-GPC4 gene cluster in one family with Simpson-Golabi-Behmel syndrome. Genomics. 1998;53 :1–11. doi: 10.1006/geno.1998.5465. [DOI] [PubMed] [Google Scholar]
- Visser R, Matsumoto N. Genetics of Sotos syndrome. Current Opin Ped. 2003;15:598–606. doi: 10.1097/00008480-200312000-00010. [DOI] [PubMed] [Google Scholar]
- Weaver DD, Graham CB, Thomas IT, et al. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly. J Pediat. 1974;84 :547–554. doi: 10.1016/s0022-3476(74)80675-x. [DOI] [PubMed] [Google Scholar]
- Weksberg R, Nishikawa J, Caluseriu O, et al. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum Mol Genet. 2001;10 :2989–3000. doi: 10.1093/hmg/10.26.2989. [DOI] [PubMed] [Google Scholar]
- Wiedemann HR. Tumours and hemihypertrophy associated with Wiedemann-Beckwith syndrome. Europ J Pediat. 1983;141 :129. [Google Scholar]
- Williams D, Gauthier DW, Maizels M. Prenatal diagnosis of Beckwith-Wiedemann syndrome. Prenat Diagn. 2005;25 :879–884. doi: 10.1002/pd.1155. [DOI] [PubMed] [Google Scholar]
- Xuan JY, Hughes-Benzie RM, MacKenzie AE. A small interstitial deletion in the GPC3 gene causes Simpson-Golabi-Behmel syndrome in a Dutch-Canadian family. J Med Genet. 1999;36 :57–58. [PMC free article] [PubMed] [Google Scholar]
- Yamashita H, Yasuhi I, Ishimaru T, et al. A case of nondiabetic macrosomia with Simpson-Golabi-Behmel syndrome: Antenatal sonographic findings. Fetal Diagn Ther. 1995;10:134–138. doi: 10.1159/000264220. [DOI] [PubMed] [Google Scholar]