

Abstract
Phosphonate natural products have proven to be a rich source of useful pharmaceutical, agricultural and biotechnology products, whereas study of their biosynthetic pathways has revealed numerous intriguing enzymes that catalyze unprecedented biochemistry. Here we review the history of phosphonate natural product discovery, highlighting technological advances that have played a key role in the recent advances in their discovery. Central to these developments has been the application of genomics, which allowed discovery and development of a global phosphonate metabolic framework to guide research efforts. This framework suggests that the future of phosphonate natural products remains bright, with many new compounds and pathways yet to be discovered.
Keywords: Phosphonate, Natural product, Antibiotic, Genome mining, Streptomyces
Introduction
Phosphonates, including both synthetic and natural products, are among one of the most widely used classes of compounds, with applications in medicine (drugs), agriculture (fertilizers and herbicides), synthetic chemistry (catalysts), industrial processes (anti-scaling, metal chelation, and water treatment), and consumer products (cosmetics and detergents) [61,39]. The diverse uses of phosphonates can be ascribed in large part to their defining chemical characteristic – the carbon-phosphorous (C-P) bond, with phosphonates containing a single C-P bond and phosphinates having either carbon-phosphorus-carbon bonds (C-P-C) or carbon-phosphorous-proton bonds (C-P-H) [39].
The diverse uses of C-P molecules has led to considerable interest in their chemistry and biochemistry. In particular, with the rising incidence of drug-resistant pathogens, there has been renewed interest in phosphonate natural products as a source of new antibiotics to mitigate this growing threat to human health. In this article we review the field of phosphonate natural products, with emphasis on how recent advances in genomics and detection methodologies are revolutionizing the discovery of new compounds and their biosynthetic pathways.
The Discovery of Phosphonate Macromolecules
The discovery of biologically produced phosphonate compounds originates not with a small molecule natural product antibiotic, but rather with the report of phosphonolipids synthesized by protozoa. Ciliatine, or 2-aminoethyl phosphonate (2-AEP) was discovered in 1959 (Figure 1) from acid-hydrolyzed extracts of protozoa in sheep rumens [22] and subsequently as the headgroup of phosphonolipids produced by many other microorganisms, animals, and even plants. Since then, phosphonoalanine and 1-hydroxy-2-aminoethylphosphonate have also been found as the headgroup of phosphonolipids [21,40]. 2-AEP and methylphosphonate are components within phosphonoglycans produced by archaea, bacteria, protozoa, and invertebrates. Additionally, both 2AEP and 2-hydroxyethyl phosphonate (2-HEP) have also been found attached to the sugars of glycosylated proteins of lower eukaryotes [21,38]. Despite their ubiquity, the biological function of phosphonate macromolecules remains an unsolved mystery.
Figure 1. Timeline of the discovered phosphonate natural products since 1959.

The biosynthetic genes for several compounds have been discovered (blue). Genome mining has thus far yielded the discovery of two new natural products (orange). Compounds that have reached clinical testing, been developed into commercial antibiotics, or biotechnological products are highlighted (red asterisks).
The Discovery of Phosphonate Natural Product Antibiotics
The majority of known small molecule phosphonate natural products isolated in the 20th century (Figure 1, Table 1) [39,16] were discovered using bioassay-guided fractionation. Accordingly, all compounds described during this time period have known bioactivities. Fosfomycin (phosphonomycin) [19,8], bialaphos (SF-1293, phosphinothricin tripeptide, PTT) [1,41], phosalacine [45,46], trialaphos [28], dehydrophos (A53868) [24], FR-900098 [43], fosmidomycin [44], the plumbemycins [47,49,48], SF-2312 [60], and fosfonochlorin [58] were originally isolated by assaying inhibition of bacterial growth. Inhibition of fungal growth was used in isolation of the fosfazinomycins [18,42] and the rhizocticins [51], while inhibition of angiotensin converting enzyme (ACE) activity was used in discovery of I5B2 [29] and K-26 [65], and prevention of seed germination in the discovery of phosphonothrixin [33,57].
Table 1. Phosphonate structure, bioactivity, year of discovery and reference.
Phosphonate natural products are listed by year of discovery. Chemical structure data is based on current knowledge of revised structures where this differs from original proposals. Asterisk next to herbicide bioactivities indicates that the compound also has known antibiotic bioactivity against E. coli in media without glutamine [28].
| Structure | Compound Name | Bioactivity | Year Discovered |
Reference |
|---|---|---|---|---|
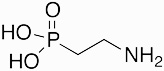 |
2-AEP | 1959 | [22] | |
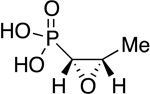 |
Fosfomycin | antibiotic | 1969 | [19] |
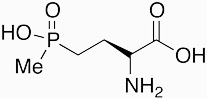 |
Phosphinothricin | herbicide | 1972 | [1] |
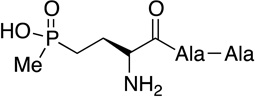 |
Bialaphos | herbicide | 1972 | [1] |
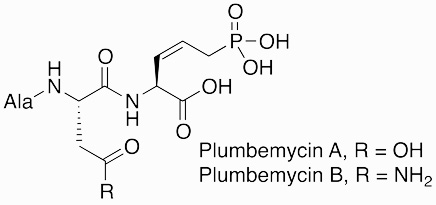 |
Plumbemycin | antibiotic | 1977 | [48] |
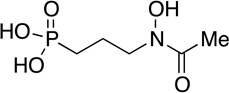 |
FR-900098 | antibiotic | 1980 | [43] |
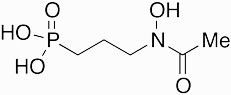 |
Fosmidomycin | antibiotic | 1980 | [44] |
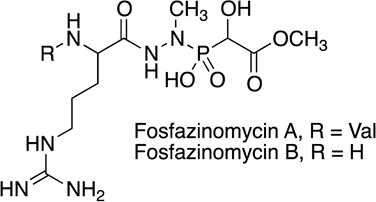 |
Fosfazinomycin | antifungal | 1983 | [42] |
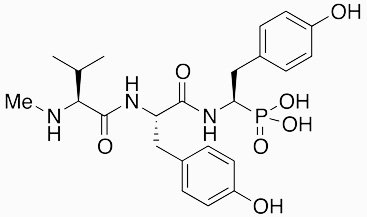 |
I5B2 | ACE inhibitor | 1984 | [29] |
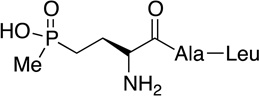 |
Phosalacine | herbicide | 1984 | [45,46] |
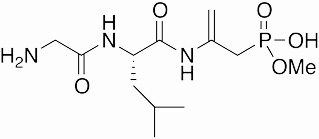 |
Dehydrophos | antibiotic | 1984 | [25,24] |
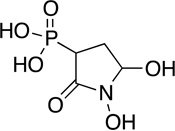 |
SF-2312 | antibiotic | 1986 | [60] |
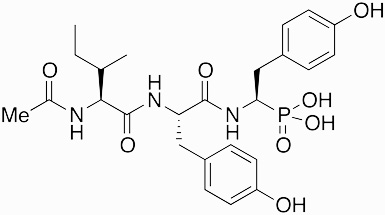 |
K-26 | ACE inhibitor | 1986 | [65] |
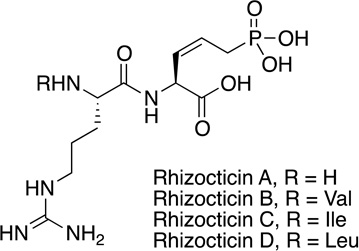 |
Rhizocticin | antibiotic | 1988 | [51] |
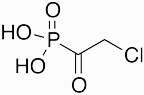 |
Fosfonochlorin | antibiotic | 1989 | [58] |
 |
Trialaphos | herbicide | 1991 | [28] |
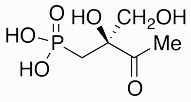 |
Phosphonothrixin | herbicide | 1995 | [57] |
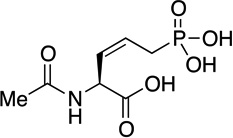 |
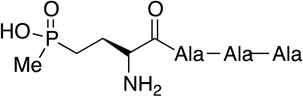
Phosacetamycin | antibiotic | 2013 | [15] |
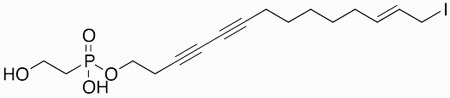 |
Phosphoiodyn A | hPPARδ agonist | 2013 | [31] |
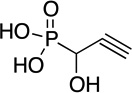 |
Cyanophos | 2013 | [9] |
The biological activity of phosphonate natural products is attributed to the chemical stability of the C-P bond coupled with chemical mimicry of key phosphate ester and anhydride metabolites. Fosfomycin, a broad-spectrum bactericide, is an analog of phosphoenolpyruvate (PEP) and inhibits UDP-N-acetylglucosamine enolpyruvyl transferase (MurA) to block peptidoglycan biosynthesis [30,37]. Similarly, fosmidomycin and FR-900098 inhibit DXP-reductoisomerase in the non-mevalonate pathway of isoprenoid biosynthesis, acting as chemical analogs of the native substrate, 2-C-methylerythritol 4-phosphate [54]. Dehydrophos is metabolized into methyl acetylphosphonate, a toxic analog of pyruvate [10]. The phosphonate peptides PTT, phosalacine, trialaphos, rhizocticins and plumbemycins are all transported into the cell and activated by proteases to release a “Trojan horse” warhead [12,34]. The non-proteinogenic amino acid (Z)-L-2-amino-5-phosphono-3-pentenoic acid (APPA) from plumbemycins and rhizocticins inhibits threonine synthase by mimicry of its natural substrate phosphohomoserine, whereas phosphinothricin from bialaphos, phosalacine, and trialaphos inhibits glutamine synthetase due to its resemblance of glutamic acid [39].
Phosphonate natural products and/or their resistance markers have been successfully commercialized into several important pharmaceutical, agricultural, and biotechnology products. Fosfomycin (Monuril®) is clinically used for the treatment of urinary tract and gastrointestinal infections. Both fosmidomycin and FR-900098 have undergone testing in clinical trials against Plasmodium, the causative agent of malaria, as their use of the non-mevalonate pathway for isoprenoid biosynthesis presents a drug target that is absent in humans [62,36,26]. Phosphinothricin (PT) is used as the active ingredient in several widely used commercial herbicide formulations sold by Bayer including Liberty®, Basta®, and Rely®. The genes encoding for bialaphos resistance (phosphinothricin acetyltransferase), originally cloned from bialaphos producing streptomycetes [63,56,59,3], have been used as selection markers in plant biology and engineering [11]. Several PT resistant varieties of canola, corn, cotton, and soybeans have been developed by Bayer and marketed as LibertyLink® crops for use in combination with their commercial PT, or glufosinate, herbicides for weed management. The commercialization of three of the 20 small molecule phosphonates shown in Figure 1 yields a success rate of 16%, two orders of magnitude higher than the 0.1% estimated for natural products as a whole [2].
The state of phosphonate natural products at the end of the 20th century
Despite this tremendous existing value and potential, less than 30 small molecule phosphonate natural products have been described in the literature (including peptide derivatives). However, the use of bioassay guided purification, rather than sensitive analytical methods for the detection of the phosphonate functional group, precluded researchers from selectively discovering new members of this natural product class. Further, many phosphonates may have been missed in screening campaigns due to the specifics of the bioassays being used. It is also likely that the chemical characteristics of the phosphonate moiety may have affected their inclusion in follow-up studies. Accordingly, most phosphonate natural products are highly water-soluble and lack sufficient hydrophobicity for extraction into partitioning organic solvents. This makes them less likely to be carried through the usual protocols for purification and characterization of molecules present in crude microbial extracts. This problem is exemplified in a passage from the introduction to the chapter on water soluble compounds in the book Natural Product Isolation: “…the purification of small water-soluble molecules is still considered to be difficult and shunned by most researchers” [52].
At the close of the 20th century, three key questions emerged that needed to be answered to revitalize the lagging field of natural product discovery: (1) How can discovery pipelines be effectively and facilely de-replicated to avoid re-purification of known compounds, (2) how many different natural products remained to be discovered and (3) which group(s) of organisms should be targeted for discovery efforts. The problem of re-discovery has become especially problematic for all classes of antibiotics, including many phosphonates. For example, after its original discovery in S. fradiae, fosfomycin was re-discovered in numerous streptomycetes and also in Pseudomonas [55,27]. The isolation of related phosphonate natural products from different strains begs the second question of whether the number of discrete scaffolds, and therefore the number of fundamentally different compounds in existence is limited. Indeed, PT, APPA, (R)-1-amino-2-(4- hydroxyphenyl)ethylphosphonic acid (AHEP; K-26/I5B2) and methyl 1-hydrophosphonoacetate (Me-HPnA; fosfazinomycin A and B) are the phosphonate moieties present in nearly half of the discovered phosphonate natural products. Also uncertain is whether actinomycetes will remain as the best source for new phosphonate natural products and whether their chemical diversity has already exhausted. Phosphonates have been purified from filamentous fungi (fosfonochlorin), bacilli (rhizocticins), and even marine sponges (phosphoiodyn A) [31], and strains from these groups could also be explored. In general, where should discovery efforts be focused to most efficiently isolate new compounds?
We have made good progress towards understanding the number of gene clusters that will be discovered via genomics. It is possible to group natural product biosynthetic gene clusters into gene cluster families (GCF) that will direct the biosynthesis of similar compounds. The conservation of GCFs between different species has been shown to vary between genera and based upon the class of natural product in question [13]. Phosphonates exist in approximately 5% of the bacterial genomes sequenced to date, a number that also applies to many of the metagenomic data sets available [66]. Recognizing the abundance of gene clusters does not address the problem of cryptic clusters, however. Many genome-based inquiries into novel natural products must first distinguish whether the many natural product gene clusters present are expressed or silent. If expression does occur under laboratory conditions, then the compounds of interest must somehow be identified out of its chemical milieu. For many classes of natural products this can be a very time consuming step, but it is a relatively straightforward aspect of phosphonate research. As will be described in the following sections, phosphonates possess a number of features that lend themselves to facile and potentially high-throughput approaches to answer each of the difficult questions posed above.
Detection of Phosphonates
Unlike the many other natural products classes (e.g. polyketides, non-ribosomally synthesized peptides, aminoglycosides, etc.) phosphonates share a simple chemical handle, observable by nuclear magnetic resonance (NMR) that allows facile detection of the molecules when they are produced. Whereas most phosphorus-containing biomolecules have 31P NMR chemical shifts between +5 to −25 ppm, all phosphonate natural products identified thus far (except phosphonoformate) have chemical shifts above 8 ppm. Thus, without any knowledge of chemical structure, exact mass, biological activity or chemical reactivity, it is possible to determine whether most phosphonates are present in a sample by a simple test. It should be noted that some non-phosphonate compounds, including cyclic phosphate esters, shift in this region, while a few phosphonates do not. Thus, we have developed two complementary methods for specific detection of phosphonic acids. The first utilizes highly sensitive mass spectrometry to create a list of putative phosphonate compounds in extracts based on ratios of negative ions after electrospray ionization with m/z 63, 79 and 97 [15]. The second method requires the molecule to have antibacterial activity and relies on an engineered E. coli strain, WM6242 that expresses the non-specific phosphonate uptake system (phnCDE) under control of a Ptac promoter. Thus, phosphonates can be deemed present when sensitivity to an extract is increased after induction of the transporter with IPTG [14]. All three approaches have been useful for detecting new molecules, characterizing the effects of genetic deletions on biosynthetic gene clusters, screening growth conditions to optimize yield, and assaying chromatography fractions.
In addition to these convenient chemical and bioassay approaches, we have developed a simple, scalable molecular biological approach to assess whether the genes needed for phosphonate biosynthesis are present. With the exception of K-26 and I5B2, all known phosphonate biosynthetic pathways have the same first step in which phosphoenolpyruvate (PEP) is reversibly converted to phosphonopyruvate (PnPy) by the enzyme PEP mutase (PepM, Figure 4). This activity was first recognized through biochemical characterization of 2-AEP biosynthesis in Tetrahymena and bialaphos production in Streptomyces hygroscopicus (Figure 2) [53,7,20]. This enzyme is a member of the isocitrate lyase superfamily, which also includes methylisocitrate lyase, 2,3-dimethylmalate lyase and phosphonpyruvate hydrolase. Although there is substantial sequence conservation between family members, a key active site motif, EDK-X5-NS, can be used to determine whether individual sequences encode bona fide PEP mutases.
Figure 4. PepM maximum-likelihood tree, early biosynthetic steps, and taxonomy.

The phylogenetic tree is the same as shown in Figure 3. Here the inner circle is colored according to the presence of phosphonopyruvate decarboxylase (Ppd), 2-AEP transaminase, phosphonoacetaldehyde reductase (PhpC) and FrbC. The outer circle is the same as in Figure 3 and shows the phylum level taxonomic designation for each strain. Sequences that correspond to known compounds are: 1, bialaphos; 2, cyanophos; 3, dehydrophos; 4, fosfazinomycin; 5, FR-900098; 6, rhizocticin; and 7, fosfomycin. The tree is rooted on the sequence for methylisocitrate lyase from E. coli, indicated with an asterisk.
Figure 2. Pathways for phosphonate natural product biosynthesis.

Genetic and biochemical characterization of phosphonate producing strains have resulted in the identification of five core biosynthetic pathways common to these natural products. PnPy and PnAA are converted into PMM, 2-keto-4-hydroxy-5-phosphonpentanoic acid (KHPTA), 2-HEP, PnAc, or 2-AEP. Dashed arrows indicate pathways newly discovered as a result of genome mining.
The use of the pepM gene as a molecular marker for phosphonate biosynthetic capacity provides a powerful tool for assessing the diversity and abundance of phosphonate biosynthesis in nature, as well as for identification of the genes needed for the synthesis of known phosphonate natural products. We have found that all known pepM sequences may be amplified with only four primer sets. The pepM genes from numerous organisms, verified in this way, were successfully used as markers to identify the gene clusters responsible for bialaphos, fosfomycin, rhizocticin, and FR-900098 biosynthesis [6,32,64,14,4]. Similarly, it is possible to predict the total number of phosphonate natural products in different environments using an amplicon sequencing approach. Recent analyses have shown that there is a direct, linear correlation between PepM sequence conservation and the associated biosynthetic gene clusters [66]. Thus, the relative similarity of two biosynthetic gene clusters, and therefore between two phosphonate biosynthetic pathways, can be predicted by examining the similarity of the PepM proteins encoded within these gene clusters. Extrapolation of these results from the observed PepM diversity in genomes, metagenomes and bacterial isolates suggests that 100–200 discrete phosphonate scaffolds await discovery. In this way, pepM as a genetic handle is not only a bioprospecting tool in the lab, but is also a potentially valuable tool for assessing the microbial diversity of an entire class of natural products. Coupled with the available chemical detection methods described above, phosphonates are an ideal system for further research into the molecular biology and ecology of natural products.
Diversity and Evolution of Phosphonate Biosynthetic Pathways
Based on the presence of pepM in sequenced genomes, synthesis of phosphonates is quite common in bacteria (270 out of 6878 genomes examined, Figure 3). The effect of horizontal gene transfer on the PepM phylogeny is quite clear, as specific taxonomic domains often appear in separate PepM lineages. Many PEP mutase sequences in this data set are linked to additional nucleotidyl transferase domains, as either N- or C-terminal fusions, which may be involved in shifting the chemical equilibrium of the PEP mutase reaction towards formation of product (see below). The N-terminal fusion appears to have evolved once and the C-terminal fusion appears to have occurred at least twice in separate branches of the PepM phylogeny. The N-terminal nucleotidyl transferase appears to have been lost in the PepM lineage present in the Burkholderia in the lower left of Figure 3. The nucleotidyl transferase domain appears to be completely absent in these lineages, as the loss of the N-terminal fusion is not accompanied by the gain of a C-terminal fusion or a separate nucleotidyl-transferase ORF.
Figure 3. PepM maximum-likelihood tree, nucleotidyl transferases and taxonomy.

A maximum-likelihood tree of full-length PepM sequences found in all bacterial genomes in NCBI as of February 2013 is shown. This tree was made using FastTreeMP with the Gamma20 option [50] and altered with the Python library ETE [23]. The inner circle next to the tree is colored based on the presence of an N- or C-terminal nucleotidyltransferase fusion with PepM, along with the presence or absence of a nucleotidyl transferase on a separate ORF. The outer circle shows the phylum level taxonomic designation for each strain. The tree is rooted on the sequence for methylisocitrate lyase from E. coli, indicated with an asterisk.
Examination of the gene neighborhoods surrounding pepM genes provides a window into the chemical diversity of the phosphonate natural product class. The chemical equilibrium for the PEP mutase reaction lies strongly towards PEP. Therefore phosphonate biosynthetic pathways always encode a thermodynamically favorable reaction that utilizes PnPy as substrate. (As noted above, the nucleotidyl transferase domain may also help drive the reaction.) In some organisms, the unfavorable production of PnPy is pulled forward through condensation with acetyl-CoA to form 2-phosphonomethylmalate (as in FR-900098 and fosfomycin biosynthesis [32,14]); however, the enzyme that most commonly fulfills this role in is PnPy decarboxylase (Ppd), which produces phosphonoacetaldehyde (PnAA, Figure 2). Following the Ppd reaction, many phosphonate biosynthetic pathways produce 2-AEP via transamination of phosphonoacetaldehyde. The transaminase that catalyzes this reaction is widespread suggesting that it is either an intermediate or the final product in many organisms, especially those outside of the actinomycetes. The majority of known small molecule phosphonates do not proceed through transamination after decarboxylation, but rather through reduction of phosphonoacetaldehyde to 2-hydroxyethylphosphonate (2-HEP), as with PhpC in PTT biosynthesis [5]. As discussed above, 2-AEP is commonly found attached to polysaccharides and as a lipid head groups and many of the gene clusters shown from Figure 4 are likely involved in the biosynthesis of these structural molecules [66]. Recent investigations into the biosynthesis of fosfazinomycin, discussed below, suggest an additional branch from PnAA to phosphonoacetate (PnAc) in this pathway. Interestingly, many gene clusters (shown as white spaces in Figure 4) lack the known thermodynamically favorable driving reactions and thus encode as-yet undiscovered biosynthetic machinery following PnPy production. Lastly, we note that all phosphonate natural products of industrial interest, with the exception of fosfomycin, are clustered within discrete clades, and all but the rhizocticins are produced by actinomycetes. Accordingly, there is a higher diversity in these biosynthetic gene clusters even when considering the first one or two steps after production of PnPy.
Recently Discovered Phosphonate Natural Products
Our group has successfully used pepM-guided genome mining in the discovery of novel phosphonate natural products from a variety of different Streptomyces strains. NMR-guided fractionation was used in the isolation of three new compounds from S. regensis strain WC-3744 [9]. From this strain, N-acetyl-2AEP, N-acetyl-1-hydroxy-2AEP were purified along with cyanophos (cyano[hydroxyl]methyl phosphonic acid), a novel cyanohydrin compound. Analysis of the phosphonate gene clusters suggests that cyanophos may have origins from 2-AEP and is likely only an intermediate in the biosynthesis of a yet to be determined final product [9].
A PCR screen for pepM also indicated the presence of a phosphonate biosynthetic gene cluster in S. atratus strain B-2808 [15]. Analysis of the fragmentation patterns of phosphonates and phosphate esters revealed a general trend where in the relative abundances of m/z 63, 79 and 97 ions were distinct between the two groups of compounds; extracts from B-2808 contained compounds with m/z ratios suggestive of phosphonates. Detection of phosphonates from concentrated crude culture extracts was further enhanced by pretreating with alkaline phosphatase and phosphodiesterase to hydrolyze phosphate esters, then precipitating free phosphates with calcium acetate, followed with enrichment by weak-anion exchange chromatography. Combining this clean-up strategy with the precursor ion scanning method and a high-resolution spectrometry instrument, phosacetamycin (N-acetyl-APPA) was identified and isolated by a mass-guided liquid chromatography approach. While the rhizocticins and plumbemycins exclusively inhibited fungi or bacteria, respectively, phosacetamycin inhibited both [15].
Most recently we have developed a stable isotope strategy to discover novel phosphonate compounds by reacting partially purified culture extracts with phosphonate O-methyltransferase DhpI (from dehydrophos biosynthesis) [35] and a mixture of SAM and CD3-SAM [17]. The chemoselective activity of DhpI enables identification by searching for pairs of compounds with an exact isotopic difference of 3.0188 Da, corresponding to SAM- and CD3-SAM-methylated phosphonates. This strategy was used to identify and purify methyl phosphonoacetate and methyl 1-hydroxyphosphonoacetate from Streptomyces sp. Strains WM6372 and XY332. Although analysis of their phosphonate gene clusters indicated the production of unknown phosphonate compounds and both molecules were undeniably new phosphonate natural products, their conspicuous similarity as a substructure of the fosfazinomycins prompted further investigations. Ultimately both strains were shown to produce phosphonates fosfazinomycin A and B. The biosynthetic genes identified from draft genomes and heterologous expression experiments provide insights into the formation of the hydrazide core and peptide formation in this structurally rare natural product [17].
Summary and Outlook
Genomics and new detection technologies developed in the past 10 years are providing both a much needed resurgence and new roadmap for continued research of this historically successful class of natural products. Even as the field of phosphonate natural products has matured significantly, many mysteries remain to be solved. The PepM independent pathways for phosphonate biosynthesis (K-26 and I5B2) have yet to be identified. Also unknown are the physiological role of some phosphonate natural products, and why certain biosynthetic pathways are highly abundant while others are rare. What is clear is that a plethora of new phosphonates, pathways, and enzymes have yet to be characterized. Genomics has unlocked a vast trove of phosphonate treasures, with many of the best discoveries sure to come in the near future.
Acknowledgements
This work was supported by the National Institute of General Medical Science of the National Institutes of Health under P01GM077596 awarded to WWM. KSJ was funded by an NIH Ruth L. Kirschstein National Research Service Award (F32GM100658) from NIGMS, and JRD by an Institute for Genomic Biology Postdoctoral Fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Baeyer E, Gugel KH, Haegele K, Hagenmaier H, Jessipow S, Koenig WA, Zaehner J. Stofwechselprodukte von Mikroorganismen 98. Phosphinothricin and phosphinothricyle-alanyl-alanin. Helv Chim Acata. 1972;55:224–239. doi: 10.1002/hlca.19720550126. [DOI] [PubMed] [Google Scholar]
- 2.Bérdy J. Thoughts and facts about antibiotics: Where we are now and where we are heading. The Journal of Antibiotics. 2012;65(8):385–395. doi: 10.1038/ja.2012.27. [DOI] [PubMed] [Google Scholar]
- 3.Block MD, Botterman J, Vandewiele M, Dockx J, Thoen C, Gossele V, Movva NR, Thompson C, Montagu MV, Leemans J. Engineering herbicide resistance in plants by expression of a detoxifying enzyme. Embo J. 1987;6(9):2513–2518. doi: 10.1002/j.1460-2075.1987.tb02537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blodgett JA, Zhang JK, Metcalf WW. Molecular cloning, sequence analysis, and heterologous expression of the phosphinothricin tripeptide biosynthetic gene cluster from Streptomyces viridochromogenes DSM 40736. Antimicrob Agents Chemother. 2005;49(1):230–240. doi: 10.1128/AAC.49.1.230-240.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blodgett JAV, Thomas PM, Li G, Velasquez JE, Donk WAvd, Kelleher NL, Metcalf WW. Unusual transformations in the biosynthesis of the antibiotic phosphinothricin tripeptide. Nat Chem Biol. 2007;3 doi: 10.1038/nchembio.2007.9. (d8f2bb67-9a19-5a0a-b099-335bae156967) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borisova SA, Circello BT, Zhang JK, van der Donk WA, Metcalf WW. Biosynthesis of rhizocticins, antifungal phosphonate oligopeptides produced by Bacillus subtilis ATCC6633. Chem Biol. 2010;17(1):28–37. doi: 10.1016/j.chembiol.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowman E, McQueney M, Barry RJ, Dunaway-Mariano D. Catalysis and thermodynamics of the phosphoenolpyruvate/phosphonopyruvate rearrangement. Entry into the phosphonate class of naturally occurring organophosphorous compounds. J Am Chem Soc. 1988;110:5575–5576. [Google Scholar]
- 8.Christensen BG, Leanza WJ, Beattie TR, Patchett AA, Arison BH, Ormond RE, Kuehl FA, Jr, Albers- Schonberg G, Jardetzky O. Phosphonomycin: structure and synthesis. Science. 1969;166(3901):123–125. doi: 10.1126/science.166.3901.123. [DOI] [PubMed] [Google Scholar]
- 9.Cioni JP, Doroghazi J, Ju K-S, Yu X, Evans BS, Metcalf WW. A cyanohydrin phosphonoate natural product from Streptomyces regensis. 2013 doi: 10.1021/np400722m. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Circello BT, Miller CG, Lee JH, van der Donk WA, Metcalf WW. The antibiotic dehydrophos is converted to a toxic pyruvate analog by peptide bond cleavage in Salmonella enterica. Antimicrob Agents Chemother. 2011;55(7):3357–3362. doi: 10.1128/AAC.01483-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D'Halluin K, De Block M, Denecke J, Janssens J, Leemans J, Reynaerts A, Botterman J. The bar gene as selectable and screenable marker in plant engineering. Methods in enzymology. 1992;216:415–426. doi: 10.1016/0076-6879(92)16038-l. [DOI] [PubMed] [Google Scholar]
- 12.Diddens H, Zahner H, Kraas E, Gohring W, Jung G. On the transport of tripeptide antibiotics in bacteria. European journal of biochemistry / FEBS. 1976;66(1):11–23. doi: 10.1111/j.1432-1033.1976.tb10420.x. [DOI] [PubMed] [Google Scholar]
- 13.Doroghazi JR, Metcalf WW. Comparative genomics of actinomycetes with a focus on natural product biosynthetic genes. BMC Genomics. 2013;14(1):611. doi: 10.1186/1471-2164-14-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eliot AC, Griffin BM, Thomas PM, Johannes TW, Kelleher NL, Zhao H, Metcalf WW. Cloning, expression, and biochemical characterization of Streptomyces rubellomurinus genes required for biosynthesis of antimalarial compound FR900098. Chem Biol. 2008;15(8):765–770. doi: 10.1016/j.chembiol.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans BS, Zhao C, Gao J, Evans CM, Ju KS, Doroghazi JR, van der Donk WA, Kelleher NL, Metcalf WW. Discovery of the antibiotic phosacetamycin via a new mass spectrometry-based method for phosphonic acid detection. ACS Chem Biol. 2013;8(5):908–913. doi: 10.1021/cb400102t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fields SC. Synthesis of natural products containing a C-P bond. Tetrahedron. 1999;55:12237–12273. [Google Scholar]
- 17.Gao J, Ju K-S, Yu X, Velasquez JE, Mukherjee S, Lee J, Zhao C, Evans BS, Doroghazi J, Metcalf WW, van der Donk WA. Use of a phosphonate methyltransferase in the identification of the fosfazinomycin biosynthetic gene cluster. 2013 doi: 10.1002/anie.201308363. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunji S, Arima K, Beppu T. Screening of antifungal antibiotics according to activities inducing morphological abnormalities. Agric Biol Chem. 1983;47:2061–2069. [Google Scholar]
- 19.Hendlin D, Stapley EO, Jackson M, Wallick H, Miller AK, Wolf FJ, Miller TW, Chaiet L, Kahan FM, Foltz EL, Woodruff HB, Mata JM, Hernandez S, Mochales S. Phosphonomycin, a new antibiotic produced by strains of streptomyces. Science. 1969;166(3901):122–123. doi: 10.1126/science.166.3901.122. [DOI] [PubMed] [Google Scholar]
- 20.Hidaka T, Mori M, Imai S, Hara O, Nagaoka K, Seto H. Studies on the biosynthesis of bialaphos (SF-1293). 9. Biochemical mechanism of C-P bond formation in bialaphos: discovery of phosphoenolpyruvate phosphomutase which catalyzes the formation of phosphonopyruvate from phosphoenolpyruvate. The Journal of antibiotics. 1989;42(3):491–494. doi: 10.7164/antibiotics.42.491. [DOI] [PubMed] [Google Scholar]
- 21.Hilderbrand RL. The role of phosphonates in living systems. CRC Press; 1983. [Google Scholar]
- 22.Horiguchi M, Kandatsu M. Isolation of 2-aminoethane phosphonic acid from rumen protozoa. Nature. 1959;184(Suppl 12):901–902. doi: 10.1038/184901b0. [DOI] [PubMed] [Google Scholar]
- 23.Huerta-Cepas J, Dopazo J, Gabaldón T. ETE: a python Environment for Tree Exploration. BMC Bioinformatics. 2010;11(1):24. doi: 10.1186/1471-2105-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hunt AH, Elzey TK. Revised structure of A53868A. The Journal of antibiotics. 1988;41(6):802. doi: 10.7164/antibiotics.41.802. [DOI] [PubMed] [Google Scholar]
- 25.Johnson R, Gordee R, Kastner R, Larsen S. Ose E Antibiotic A53868 and Process for Production Thereof. 2,127,413. Eli Lilly: Chem. Abstr; UK Patent. 1984:88837. 1984.
- 26.Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Turbachova I, Eberl M, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science. 1999;285(5433):1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- 27.Katayama N, Tsubotani S, Nozaki Y, Harada S, Ono H. Fosfadecin and fosfocytocin, new nucleotide antibiotics produced by bacteria. The Journal of antibiotics. 1990;43(3):238–246. doi: 10.7164/antibiotics.43.238. [DOI] [PubMed] [Google Scholar]
- 28.Kato H, Nagayama K, Abe H, Kobayashi R, Ishihara E. Isolation, structure and biological activity of trialaphos. Agric Biol Chem. 1991;55:1133–1134. [Google Scholar]
- 29.Kido Y, Hamakado T, Anno M, Miyagawa E, Motoki Y, Wakamiya T, Shiba T. Isolation and characterization of I5B2, a new phosphorus containing inhibitor of angiotensin I converting enzyme produced by Actinomadura sp. The Journal of antibiotics. 1984;37(9):965–969. doi: 10.7164/antibiotics.37.965. [DOI] [PubMed] [Google Scholar]
- 30.Kim DH, Lees WJ, Kempsell KE, Lane WS, Duncan K, Walsh CT. Characterization of a Cys115 to Asp substitution in the Escherichia coli cell wall biosynthetic enzyme UDP-GlcNAc enolpyruvyl transferase (MurA) that confers resistance to inactivation by the antibiotic fosfomycin. Biochemistry. 1996;35(15):4923–4928. doi: 10.1021/bi952937w. [DOI] [PubMed] [Google Scholar]
- 31.Kim H, Chin J, Choi H, Baek K, Lee TG, Park SE, Wang W, Hahn D, Yang I, Lee J, Mun B, Ekins M, Nam SJ, Kang H. Phosphoiodyns A and B, unique phosphorus-containing iodinated polyacetylenes from a Korean sponge Placospongia sp. Org Lett. 2013;15(1):100–103. doi: 10.1021/ol3031318. [DOI] [PubMed] [Google Scholar]
- 32.Kim SY, Ju KS, Metcalf WW, Evans BS, Kuzuyama T, van der Donk WA. Different biosynthetic pathways to fosfomycin in Pseudomonas syringae and Streptomyces species. Antimicrob Agents Chemother. 2012;56(8):4175–4183. doi: 10.1128/AAC.06478-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kimura T, Nakamura K, Takahashi E. Phosphonothrixin, a novel herbicidal antibiotic produced by Saccharothrix sp. ST-888. II. Structure determination. The Journal of antibiotics. 1995;48(10):1130–1133. doi: 10.7164/antibiotics.48.1130. [DOI] [PubMed] [Google Scholar]
- 34.Laber B, Lindell SD, Pohlenz HD. Inactivation of Escherichia coli threonine synthase by DL-Z-2- amino-5-phosphono-3-pentenoic acid. Archives of microbiology. 1994;161(5):400–403. doi: 10.1007/BF00288949. [DOI] [PubMed] [Google Scholar]
- 35.Lee JH, Bae B, Kuemin M, Circello BT, Metcalf WW, Nair SK, van der Donk WA. Characterization and structure of DhpI, a phosphonate O-methyltransferase involved in dehydrophos biosynthesis. Proc Natl Acad Sci U S A. 2010;107(41):17557–17562. doi: 10.1073/pnas.1006848107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lell B, Ruangweerayut R, Wiesner J, Missinou MA, Schindler A, Baranek T, Hintz M, Hutchinson D, Jomaa H, Kremsner PG. Fosmidomycin, a novel chemotherapeutic agent for malaria. Antimicrob Agents Chemother. 2003;47(2):735–738. doi: 10.1128/AAC.47.2.735-738.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marquardt JL, Brown ED, Lane WS, Haley TM, Ichikawa Y, Wong CH, Walsh CT. Kinetics, stoichiometry, and identification of the reactive thiolate in the inactivation of UDP-GlcNAc enolpyruvoyl transferase by the antibiotic fosfomycin. Biochemistry. 1994;33(35):10646–10651. doi: 10.1021/bi00201a011. [DOI] [PubMed] [Google Scholar]
- 38.Metcalf WW, Griffin BM, Cicchillo RM, Gao J, Janga SC, Cooke HA, Circello BT, Evans BS, Martens- Habbena W, Stahl DA, van der Donk WA. Synthesis of methylphosphonic acid by marine microbes: a source for methane in the aerobic ocean. Science. 2012;337(6098):1104–1107. doi: 10.1126/science.1219875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Metcalf WW, van der Donk WA. Biosynthesis of phosphonic and phosphinic acid natural products. Annu Rev Biochem. 2009;78:65–94. doi: 10.1146/annurev.biochem.78.091707.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moschidis MC. Phosphonolipids. Prog Lipid Res. 1985;23:223–246. doi: 10.1016/0163-7827(84)90012-2. [DOI] [PubMed] [Google Scholar]
- 41.Ogawa H, Tsuruoka T, Inouye S, Niida T. Studies on a new antibiotic SF-1293. Sci Rep Meiji Seika Kaisha. 1973;13:42–48. [Google Scholar]
- 42.Ogita T, Gunji S, Fukawa Y, Terahara A, Kinoshita T, Nagaki H. The structures of fosfazinomycins A and B. Tetrahedron Letters. 1983;24:2283–2286. [Google Scholar]
- 43.Okuhara M, Kuroda Y, Goto T, Okamoto M, Terano H, Kohsaka M, Aoki H, Imanaka H. Studies on new phosphonic acid antibiotics. I. FR-900098, isolation and characterization. The Journal of antibiotics. 1980;33(1):13–17. doi: 10.7164/antibiotics.33.13. [DOI] [PubMed] [Google Scholar]
- 44.Okuhara M, Kuroda Y, Goto T, Okamoto M, Terano H, Kohsaka M, Aoki H, Imanaka H. Studies on new phosphonic acid antibiotics. III. Isolation and characterization of FR-31564, FR-32863 and FR-33289. The Journal of antibiotics. 1980;33(1):24–28. doi: 10.7164/antibiotics.33.24. [DOI] [PubMed] [Google Scholar]
- 45.Omura S, Hinotozawa K, Imamura N, Murata M. The structure of phosalacine, a new herbicidal antibiotic containing phosphinothricin. The Journal of antibiotics. 1984;37(8):939–940. doi: 10.7164/antibiotics.37.939. [DOI] [PubMed] [Google Scholar]
- 46.Omura S, Murata M, Hanaki H, Hinotozawa K, Oiwa R, Tanaka H. Phosalacine, a new herbicidal antibiotic containing phosphinothricin. Fermentation, isolation, biological activity and mechanism of action. The Journal of antibiotics. 1984;37(8):829–835. doi: 10.7164/antibiotics.37.829. [DOI] [PubMed] [Google Scholar]
- 47.Park BK, Hirota A, Sakai H. 2-Amino-5-phosphono-3-pentenoic acid, a new amino acid from N- 1409 substrance, an antagonist of threonine. Agric Biol Chem. 1976;40(1905-1906) [Google Scholar]
- 48.Park BK, Hirota A, Sakai H. Structure of plumbemycin A and B, antagonists of L-threonine from Streptomyces plumbeus. Agric Biol Chem. 1977;41:573–579. [Google Scholar]
- 49.Park BK, Hirota A, Sakai H. Studies on new antimetabolite N-1409. Agric Biol Chem. 1977;41:161–167. [Google Scholar]
- 50.Price MN, Dehal PS, Arkin AP. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5(3):e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rapp C, Jung G, Kugler M, Loeffler W. Rhizocticins - new phosphono-oligopeptides with antifungal activity. Liebigs Annalen der Chemie. 1988:655–661. [Google Scholar]
- 52.Sarker SD, Latif Z, Gray AI. Natural products isolation, vol 20. Springer; 2005. [Google Scholar]
- 53.Seidel HM, Freeman S, Seto H, Knowles JR. Phosphonate biosynthesis: isolation of the enzyme responsible for the formation of a carbon-phosphorus bond. Nature. 1988;335(6189):457–458. doi: 10.1038/335457a0. [DOI] [PubMed] [Google Scholar]
- 54.Shigi Y. Inhibition of bacterial isoprenoid synthesis by fosmidomycin, a phosphonic-acid antibiotic. J Antimicrob Chemother. 1989;24:131–145. doi: 10.1093/jac/24.2.131. [DOI] [PubMed] [Google Scholar]
- 55.Shoji J, Kato T, Hinoo H, Hattori T, Hirooka K, Matsumoto K, Tanimoto T, Kondo E. Production of fosfomycin (phosphonomycin) by Pseudomonas syringae. The Journal of antibiotics. 1986;39(7):1011–1012. doi: 10.7164/antibiotics.39.1011. [DOI] [PubMed] [Google Scholar]
- 56.Strauch E, Wohlleben W, Puhler A. Cloning of a phosphinothricin N-acetyltransferase gene from Streptomyces viridochromogenes Tu494 and its expression in Streptomyces lividans and Escherichia coli. Gene. 1988;63(1):65–74. doi: 10.1016/0378-1119(88)90546-x. [DOI] [PubMed] [Google Scholar]
- 57.Takahashi E, Kimura T, Nakamura K, Arahira M, Iida M. Phosphonothrixin, a novel herbicidal antibiotic produced by Saccharothrix sp. ST-888. I. Taxonomy, fermentation, isolation and biological properties. The Journal of antibiotics. 1995;48(10):1124–1129. doi: 10.7164/antibiotics.48.1124. [DOI] [PubMed] [Google Scholar]
- 58.Takeuchi M, Nakajima M, Ogita T, Inukai M, Kodama K, Furuya K, Nagaki H, Haneishi T. Fosfonochlorin, a new antibiotic with spheroplast forming activity. The Journal of antibiotics. 1989;42(2):198–205. doi: 10.7164/antibiotics.42.198. [DOI] [PubMed] [Google Scholar]
- 59.Thompson CJ, Movva NR, Tizard R, Crameri R, Davies JE, Lauwereys M, Botterman J. Characterization of the herbicide-resistance gene bar from Streptomyces hygroscopicus. Embo J. 1987;6(9):2519–2523. doi: 10.1002/j.1460-2075.1987.tb02538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watanabe H, Yoshida J, Tanaka E, Ito M, Miyadoh S, Shomura T. Studies on a new phosphonic acid antibiotic, SF-2312. Sci Rep Meiji Seika Kaisha. 1986;25:12–17. [Google Scholar]
- 61.Widler L, Jahnke W, Green JR. The chemistry of bisphosphonates: from antiscaling agents to clinical therapeutics. Anticancer Agents Med Chem. 2012;12(2):95–101. doi: 10.2174/187152012799014959. [DOI] [PubMed] [Google Scholar]
- 62.Wiesner J, Borrmann S, Jomaa H. Fosmidomycin for the treatment of malaria. Parasitol Res. 2003;90(Suppl 2):S71–S76. doi: 10.1007/s00436-002-0770-9. [DOI] [PubMed] [Google Scholar]
- 63.Wohlleben W, Arnold W, Broer I, Hillemann D, Strauch E, Puhler A. Nucleotide sequence of the phosphinothricin N-acetyltransferase gene from Streptomyces viridochromogenes Tu494 and its expression in Nicotiana tabacum. Gene. 1988;70(1):25–37. doi: 10.1016/0378-1119(88)90101-1. [DOI] [PubMed] [Google Scholar]
- 64.Woodyer RD, Shao Z, Thomas PM, Kelleher NL, Blodgett JA, Metcalf WW, van der Donk WA, Zhao H. Heterologous production of fosfomycin and identification of the minimal biosynthetic gene cluster. Chem Biol. 2006;13(11):1171–1182. doi: 10.1016/j.chembiol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 65.Yamato M, Koguchi T, Okachi R, Yamada K, Nakayama K, Kase H, Karasawa A, Shuto K. K-26, a novel inhibitor of angiotensin I converting enzyme produced by an actinomycete K-26. The Journal of antibiotics. 1986;39(1):44–52. doi: 10.7164/antibiotics.39.44. [DOI] [PubMed] [Google Scholar]
- 66.Yu X, Doroghazi JR, Janga SC, Zhang JK, Circello BT, Griffin BM, Metcalf WW. The unappreciated role of phosphonates in nature. 2013 doi: 10.1073/pnas.1315107110. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]