

Abstract
Biliary excretion is one of the main elimination pathways for drugs and/or their metabolites. Therefore, an insight into the structural profile of cholephilic compounds through accurate modelling of the biliary excretion is important for the estimation of clinical pharmacokinetics in early stages of drug discovery. The aim of this study was to develop quantitative structure-activity relationships as computational tools for the estimation of biliary excretion and identification of the molecular properties controlling this process. The study used percentage of dose excreted intact into bile measured in vivo in rat for a diverse dataset of 217 compounds. Statistical techniques were multiple linear regression analysis, regression trees, random forest and boosted trees. A simple regression tree model generated using the CART algorithm was the most accurate in the estimation of the percentage of bile excretion of compounds, and this outperformed the more sophisticated boosted trees and random forest techniques. Analysis of the outliers indicated that the models perform best when lipophilicity is not too extreme (log P < 5.35) and for compounds with molecular weight above 280 Da. Molecular descriptors selected by all these models including the top ten incorporated in boosted trees and random forest indicated a higher biliary excretion for relatively hydrophilic compounds especially if they are anionic or cationic, and have a large molecular size. A statistically validated molecular weight threshold for potentially significant biliary excretion was above 348 Da.
Electronic supplementary material
The online version of this article (doi:10.1208/s12248-013-9541-z) contains supplementary material, which is available to authorized users.
KEY WORDS: bile, biliary excretion, CART, pharmacokinetic, QSAR
INTRODUCTION
Biliary excretion is an important route for the elimination of some drugs and their metabolites (1). Although the liver is generally identified with its role in metabolism, one of the most important functions of the liver is formation of bile which is then stored in the gallbladder and discharged into the duodenum upon ingestion of food, with bile carrying also cholephilic xenobiotics. Bile which is a composition of bile acids and other components such as phospholipids, bilirubin and cholesterol is formed in the hepatocytes and is actively discharged across the canalicular membrane into canaliculus (2). Once bile is released into the intestine, some metabolites and unchanged drugs continue their way of elimination through the faeces. Others, for example lipid-soluble drugs, are reabsorbed from the intestine and move to the systemic circulation (2). This enterohepatic circulation affects pharmacokinetics by keeping the plasma concentration of drugs high (1). Enterohepatic cycling and biliary elimination can continue until the compound is ultimately eliminated from the body by faecal or renal excretion or metabolism.
Uptake from sinusoidal blood and then secretion of bile salts across the canalicular hepatocyte membrane are the major factors controlling the rate of bile secretion. Figure 1 illustrates a schematic representation of some of the important transporters that are involved in this process. Basolateral bile salt uptake is driven through the Na+-dependent and Na+-independent uptake systems (3). The main sodium-dependent bile salt transporters are Na+-taurocholate co-transporting polypeptides (human and rat). On the other hand, the Na+-independent uptake of bile salts cannot be attributed to the function of a single transport system and several carrier systems have been implicated including sulphate/anion exchanger, dicarboxylate/anion exchanger and OH−/cholate exchanger. In rats, the organic anion transporting polypeptides (Oatp1, Oatp2 and Oatp4) have been indicated as the main sodium-independent uptake proteins (3). The organic cation and organic anion transporters (OCT and OAT, respectively) also play important roles in the initial sinusoidal influx of drugs into hepatocytes (4). These transporters have wide substrate specificities for a range of exogenous and endogenous substrates (5). OCT1 can be found abundantly in hepatocytes and may be seen as the most important transporter for distribution of cationic compounds into the liver from sinusoidal membrane (6).
Fig. 1.
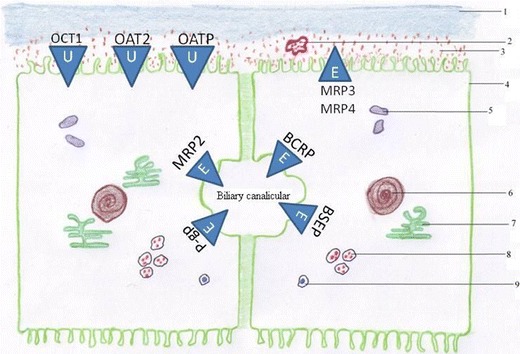
The cartoon depicts substrate transport processes in the hepatocyte including sinusoidal and canalicular proteins efflux (E) and uptake (U) transport of drugs/drug-likes and their metabolites. 1 Sinusoidal membrane, 2 Ito cell, 3 space of Disse, 4 hepatocyte, 5 mitochondria, 6 nucleus, 7 endoplasmic reticulum, 8 lysosomes, 9 peroxisome
Canalicular bile secretion is an osmotic process in which active excretion of organic solutes into the bile canaliculus is the main driving force for the passive inflow of water, electrolytes and nonelectrolytes from hepatocytes (7). While products of the multidrug resistance gene family (Mdr), namely bile salt export pumps, Bsep (rat) and BSEP (human), transport monovalent bile salts (2), excretion of non-bile salt organic anions and divalent sulphate or glucuronide bile salts is carried mainly by the multidrug resistance protein 2 (MRP2). Bile salt export pump has a limited role in drug excretion. However, drug inhibition of this pump can lead to hepatotoxicity (8). Another member of this family (P-glycoprotein), also known as multidrug resistance protein 1, actively effluxes xenobiotics into the bile (9). Breast cancer resistance protein (BCRP/ABCG2) is also involved in the transport of a range of drugs. For example, nitrofurantoin has a very high biliary excretion predominantly mediated by BCRP (10). Other basolateral isoforms of the multidrug resistance-associated protein, MRP4 and MRP3, provide alternative routes for the elimination of organic anions from hepatocytes into the systemic circulation (3).
Properties of the chemical structure as well as the characteristics of the liver such as specific active transport sites within the liver cell membranes are the main factors which determine the elimination of xenobiotics via the biliary tract (2). Despite the various transport systems involved in the biliary elimination of xenobiotics, there has been a number of attempts to identify common molecular features of highly excreted compounds. Molecular weight (MW) has been suggested as an important factor in biliary excretion levels of compounds. Anionic compounds with the MW higher than 325 ± 50 kDa in rats, 400 ± 50 kDa in guinea pigs, 475 ± 50 kDa in rabbits and 500 ± 50 kDa in human have been suggested as good candidates for biliary excretion (11). Most compounds with lower molecular weights are quickly cleared through the kidneys and are not excreted in the bile (12). Bile is rich in endogenous organic anionic substrates (e.g., steroid hormones), organic cations (such as quaternary ammonium), bilirubin and bile acids (2). Moreover, excretion route of anionic xenobiotics and some antibacterials is through the bile (13). Principally, for organic cationic compounds, biliary elimination depends on the molecular volume (14), lipophilicity of the compound and the number of cationic groups (15).
Biliary excretion has major significance in determining the pharmacokinetic profiles of drugs. In several disease states, the excretion of drugs through bile is affected and toxicities may arise (1,2). Knowledge of biliary excretion levels of compounds can help in identifying any possible mechanisms of hepatobiliary toxicity and potential drug–drug interactions. Therefore, an insight into the structural profile of cholephilic compounds through accurate modelling of the biliary excretion is important for predicting clinical pharmacokinetics. This is of a particular value during earlier stages of drug discovery where low-cost estimation procedures are required. Quantitative structure-activity relationships (QSARs) employ data mining techniques to explore the relationships between biological properties of interest, e.g. pharmacokinetic parameters of drugs, and the properties of the molecular structures (16). Recently, a QSAR model developed using 2D molecular descriptors showed good prediction ability for a set of literature biliary excretion data measured under the same experimental model (17). However, re-evaluation of this simple model showed that the statistical significance of the model is lost when it is used for the prediction of a wide set of external compounds (18), suggesting that hepatobiliary excretion cannot be captured by simple physicochemical descriptors when examining chemically dissimilar compounds. Unfortunately, availability of in vivo biliary excretion data which is necessary for modelling is very limited. Yang et al. (19) have recently compiled a big dataset of percentage of dose eliminated in the bile in rats and humans. This offers an excellent resource for a detailed study on the structural determinants for high biliary excretion. Using this dataset, Yang and co-workers suggested a MW threshold of 400 Da for anions in rats and 475 Da for anions in humans. They also developed linear regression models for human and rat. The aim of this study was to use an expanded dataset and incorporate non-linear methods to develop statistically valid QSAR models. Specifically, classification and regression tree (CART) is a flexible and yet simple and interpretable technique with embedded feature selection that selects the most significant molecular descriptor for partitioning the data into smaller subsets of similar observations (20). This rule-based technique is a decision tree that splits the data in a recursive manner until the subset has all the same value of the target (dependent) variable, or when no gain in the prediction accuracy is achievable by further splitting. In this study, we aimed at using regression trees and two ensemble methods that construct many such decision trees and return the consensus prediction by the trees, namely random forest and boosted trees. The prediction accuracy of the models and the molecular descriptors selected by these methods were compared in order to clarify the structural elements controlling the biliary excretion. Moreover, regression trees were used to examine the significance of molecular weight and presence of carboxylic acid groups and to find the statistically significant threshold values. In this case, regression trees are useful since they can be used interactively so that a molecular descriptor of choice can be incorporated at any split level and the analysis may determine the statistically significant threshold value of the descriptor for splitting the data.
METHODS
Dataset
The biliary excretion dataset collated by Yang et al. (19), available at http://www.acsu.buffalo.edu/~memorris/Suppl%20Data_Rats_052909_pdb.pdf, plus some additional data from the original literature were used in this study (see supplementary material I for the dataset and the reference list). It consists of in vivo biliary excretion expressed as the percentage of dose excreted as the parent compound intact through the bile (BE%) for 217 compounds in rat after iv or intraperitoneal administration of the compound. The compounds are from different chemical classes such as bile acids, statins, dyes, penicillins and cephalosporins, macrolide antibiotics, quinolone antibiotics, NSAIDs, thrombin inhibitors, analgesics, anti-cancer drugs such as doxorubicin, folates, peptides, anti-HIV agents, quaternary ammoniums, sulphanilamide and arylaminosulphonic acids.
Molecular Descriptors
Molecular descriptors were calculated for the 217 compounds using ACD Labs/logD suite version 12, TSAR 3D version 3.3 (Accelryl Inc), molecular operation environment (MOE) version 2011.10 (Chemical Computing Group) and Symyx QSAR software (Accelryl Inc). The fractions of ionised compounds at pH 7.4 as acid (fiA), as base (fiB) or for zwitterionic compounds ionised as acid and base (fiAB) and fraction unionised (fU) were calculated using Eqs. 1 to 4 and used as additional molecular descriptors as follows:
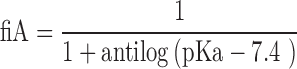 |
1 |
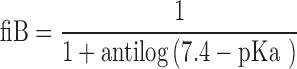 |
2 |
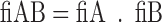 |
3 |
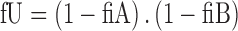 |
4 |
In Eqs. 1 and 2, pKa was the most acidic and the most basic, respectively, which were obtained from ACD Labs pKa database and, in case the experimental pKa was not available, it was calculated by the software.
In MOE, following the wash procedure which removed salt forms, energy minimization was carried in order to calculate atomic coordinates corresponding to the local minimum (the low-energy conformation). Thereafter, self-consistent field calculations were performed, and this latter energy-minimised structure was used for the calculation of all the molecular descriptors. Before building the models, the molecular descriptors were checked to find and discard those columns containing more than 98% constant values or more than 28 (out of 217) missing values. The total number of descriptors used in all statistical analyses was 387.
Model Development and Validation
Several linear and non-linear methods were used for the QSAR model development. The compounds were divided into an external validation set and a training data. Models were developed using training set compounds and assessed using internal and external validation sets. To divide the compounds, they were ordered according to BE%, and from every set of five compounds, four were allocated into the training and one into the external validation set randomly. In this way, training data consisted of 168 compounds and the external validation set consisted of 40 compounds. For the analytical methods that required parameter optimization, a fraction of training set compounds were randomly assigned into internal validation set, or alternatively, cross validation was used if the option was available in the statistical software. STATISTICA Data Miner was the software used for statistical analysis. For the internal validation set, where applicable, the risk estimate and standard error were calculated in STATISTICA software and used as the performance indicators. Risk estimate is calculated as the proportion of residual variance incorrectly estimated by the model. Standard error measures the error of the prediction.
Mean absolute error was used to assess the accuracy of prediction of biliary excretion using the selected models (Eq. 5).
 |
5 |
In Eq. 5, ‘observed’ refers to the log percentage of intact dose excreted into the bile from in vivo studies (LogBE%), ‘predicted’ is the predicted LogBE% by the QSAR models and N is the number of compounds. In all statistical analyses, the logarithm of percentage dose excreted (LogBE%) was used in the analysis instead of BE%. This was due to the normal distribution of LogBE% as indicated by the skewness comparison with BE%.
Stepwise Regression Analysis
Minitab Statistical Software version 16 was used for the development of multiple linear regression (MLR) models. In stepwise regression analysis, LogBE% was the dependant variable and all the molecular descriptors were the independent variables. In all regression analyses, a P value of less than 0.05 was considered to be statistically significant for variables. Values for ‘alpha to enter’ and ‘alpha to remove’ were set to 0.05.
Classification and Regression Trees
Classification and regression trees (C&RT) routine in STATISTICA version 11 (StatSoft Inc) was used to develop regression trees (RTs). The analysis builds an optimal tree structure to predict continuous dependent variables via V-fold cross-validation. In C&RT analysis, LogBE% was the dependent variable and the predictors were selected by this statistical analysis from all the molecular descriptors provided. The size of a tree in C&RT analysis is an important issue, since an unreasonably big tree can lead to overfitting and can make the interpretation of the results more difficult. Several stopping criteria were examined, including the default settings in STATISTICA. The default stopping criteria were minimum number of cases of 21 and the maximum number of nodes set to 100. The default V value of 10 was used in the V-fold cross-validation, and the risk-estimate was used to check the reliability of the resulting RTs.
Boosted Trees
Boosted trees analysis in STATISTICA generates a series of very simple boosting regression trees (BT) where each successive tree is built for the prediction of residuals of the preceding tree. Each of these trees has weak predictive accuracy, but using the weak predictors together can create a strong predictor (21). The default values for learning rate, the number of additive terms (number of trees), random test data proportion (fraction of data points in testing pool) and subsample proportion were 0.1, 200, 0.2 and 0.5, respectively. Various subsample proportions of 0.4, 0.45, 0.50, 0.55 and 0.60 were examined in combination with the learning rates of 0.1 and 0.05. The best two models were selected based on the performance indicators for the internal validation set. The seed for random number generation that controls which cases are selected in sampling was set to one. The maximum number of nodes was set to three, which means that each tree will have just one binary split.
Random Forest
A random forest (RF) model is an ensemble of tree predictors such that each tree depends on the values of a random vector (a random selection of molecular descriptors and training set compounds) sampled independently. The method builds a series of simple trees where the predictions are taken to be the average of the predictions of all the trees (22). Various subsample proportions of 0.40, 0.45, 0.50, 0.55 and 0.60 were examined, while the number of predictors (to be randomly considered at each node) was nine. Different numbers of trees were tested at 20, 50, 80, 100 and 200. The random test data proportion was 0.2 for the internal validation. The default settings were used for stopping conditions including minimum number of cases, maximum number of levels, minimum number in child node and the maximum number of nodes of 6, 10, 5 and 100, respectively. The best model was selected based on the estimation error for the internal test data.
RESULTS
A total of 387 2D (e.g. kappa shape indexes, molecular connectivity indexes and electrotopological state indexes) and 3D molecular descriptors were used for the QSAR model development. Out of 217 compounds in the rat biliary excretion dataset, 9 compounds had excretion rate of 0%, and hence LogBE% could not be calculated for them; 168 compounds in the training set were employed for the model development and the remaining 40 compounds served as the external validation set. The method of data allocation into training and test sets outlined above ensured that a similar biliary excretion and molecular property spaces were covered by both the training and the validation sets. BE% values ranged between 0.048 and 100 with mean LogBE% values for the training and validation sets at 1.04 and 1.01, respectively. LogP was between −3.44 and 18.8 for the training set, and −3.17 and 7.83 for the validation set with similar mean values of 1.81 and 1.83, respectively. Molecular weights of the compounds were between 122 and 1,215 Da for the training set and 94 and 1,255 Da for the validation set, with mean values of 457 and 390, respectively. Scores plot from principle component analysis using all the molecular descriptors also indicates similar chemistry space for the two sets (Figure S1 in supplementary material II).
Regression Models
Stepwise regression analysis using in vivo rat biliary excretion data as the dependant variable resulted in the MLR model below in which the number of molecular descriptors is limited to eight. The statistical terms of the equation are N the number of compounds, R2 the correlation coefficient, S the standard deviation and F Fisher’s statistics and the P value. Observed versus calculated LogBE% by this equation has been plotted (Figure S2 in supplementary material II), with training and validation sets identified in the plot.
MLR model
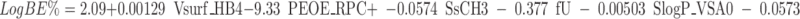 |
6 |
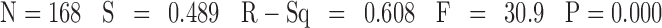 |
Molecular descriptors of this equation are not intercorrelated (R2 < 0.4). Table I gives a brief description of molecular descriptors used in this model. Vsurf_HB4 is the first molecular descriptor selected by the analysis and it indicates that compounds with high H-bond donor capacity have higher biliary excretion level. AM1_dipole (dipole moment) is the other polarity descriptor which has a positive effect. On the other hand, the equation shows that drugs with greater relative positive partial charge (PEOE_RPC+) have lower biliary excretion. The value of this descriptor is large for small acidic molecules such as benzoic acid and salicylic acid, and therefore the small size of such compounds may be the reason for the reduced biliary excretion. In this equation, fU with a negative coefficient indicates that compounds with higher unionised fraction at pH 7.4 have lower biliary excretion. In other words, although according to fU, acidity and basicity (dissociation in general) increase the biliary excretion of compounds, this is true only for large dissociated molecules. The positive effect of polarity and dissociation on biliary excretion is in agreement with the literature, where for example polar surface area (18) and an acidity indicator (17,23) have been included in linear QSAR models. Also according to this equation, compounds containing many methyl groups (SsCH3) and those that are highly branched containing >CH− groups (SsssCH) have lower biliary excretion. In this equation, SlogP_VSA0 shows the negative impact of the presence of atoms with LogP(o/w) contribution of less than or equal to −0.4. SddssS_acnt indicates the direct effect of sulphate or sulphonamide groups. Sulphate and sulphonamide groups are found in sulphonamide drugs such as succinylsulphathiazole, dyes such as methyl orange and sulphate conjugates such as estrone 3-sulphate which may be substrates of MRP2 or BCRP (24).
Table I.
A Brief Description of the Most Important Molecular Descriptors Selected and Used by the Models
| Descriptor | Model | Description |
|---|---|---|
| a_acc | RF | Number of H-bond acceptor atoms |
| a_hyd | BT (1) | Number of hydrophobic atoms |
| AM1_dipole | MLR, RT (1) | Dipole moment calculated using AM1 Hamiltonian |
| BCUT_PEOE_0 | RF | The BCUT descriptor calculated from the eigenvalues of a modified adjacency matrix. The resulting eigenvalues are sorted and the smallest, 1/3-ile, 2/3-ile and largest eigenvalues are reported, in this case the 2/3-ile. The diagonal takes the value of the PEOE partial charges. |
| CASA- | RT (3) | Negative charge weighted surface area, ASA- times max {qi < 0} |
| chi1 | RF | First order molecular connectivity index |
| COOH | RT (3) | Indicator variable for the presence of carboxylic acid group in the molecular structure |
| Docking energy (MOE) | RF | Docking score (kcal/mol) for enzyme-ligand docking of the compounds into the active site of mouse P-glycoprotein from PDB calculated using MOE software |
| FASA_H | RT (1) | Fractional ASA_H calculated (water accessible surface area of all hydrophobic atoms) as ASA_H/ASA |
| FCASA- | RT (2) | Fractional CASA- calculated as CASA-/ASA |
| fU | MLR, RT (1) | Fractions of compounds unionised. |
| GCUT_SLOGP_1 | RT (1) | The GCUT descriptors are calculated from the eigenvalues of a modified graph distance adjacency matrix. Each ij entry of the adjacency matrix takes the value 1/sqr(dij) where dij is the (modified) graph distance between atoms i and j. The resulting eigenvalues are sorted and the smallest, 1/3-ile, 2/3-ile and largest eigenvalues are reported. The diagonal takes the value of the atomic contribution to logP. |
| Kier2 | BT (1), BT (2) | Second order kappa shape index: (n−1)2/m2 |
| Kier3 | BT (1), BT (2) | Third order kappa shape index: (n−1)2/m2 |
| KierA1 | RT (2) | First order alpha modified shape index: s (s−1)2/m2 where s = n + a |
| KierA3 | BT (1), BT (2) | Third order alpha modified shape index: (n−1) (n−3)2/p3 2 for odd n, and (n−3) (n−2)2/p3 2 for even n where s = n + a |
| LogD (5.5) | BT (2) | Logarithm of distribution coefficient D of a compound between octanol and buffer layers at pH value 5.5 |
| LogD (6.5) | RT (1), RT (2), BT (1), BT (2) | Logarithm of distribution coefficient D of a compound between octanol and buffer layers at pH value 6.5 |
| LogD (7.4) | BT (1), BT (2) | Logarithm of distribution coefficient D of a compound between octanol and buffer layers at pH value 7.4 |
| LogD (10) | RT (3), BT (2) | Logarithm of distribution coefficient D of a compound between octanol and buffer layers at pH value 10 |
| MW | RT (2) RF | The molecular weight |
| N ratio | RT (1) | The weight ratio of nitrogen atoms in the molecule |
| PEOE_PC- | RT (3) | Total negative partial charge |
| PEOE_RPC+ | MLR, BT (2) | Relative positive partial charge: the largest positive atomic charge divided by the sum of the positive partial charges |
| PEOE_VSA_NEG | RT (2) | Total negative van der Waals surface area |
| PEOE_VSA-0 | RT (1) | Van der Waals surface area of atoms with atomic charge in the range (−0.05, 0.00) |
| PEOE_VSA_FPPOS | RF | Fractional positive polar van der Waals surface area. This is the sum of the VDW surface area such that partial charge of atom is greater than 0.2 |
| PEOE_VSA_HYD | BT (1), BT (2) | Total hydrophobic van der Waals surface area. This is the sum of the van der Waals surface area such that absolute value of atomic charge is less than or equal to 0.2. |
| Q_PC+ | RF | Total positive partial charge: the sum of the positive partial charge of atoms in the molecule |
| SddssS_acnt | MLR | Count of all sulphur atoms (ddssS) E-state indexes in molecule |
| SlogP_VSA0 | MLR | Sum of approximate accessible van der Waals surface area for atoms with atomic contribution to logP(o/w) of equal or less than −0.4 |
| SMR_VSA7 | RT (2) | Sum of approximate accessible van der Waals surface area for atoms with atomic contribution to molar refractivity of R i > 0.56 |
| SsCH3 | MLR | Atom type electrotopological state index (sum of the E-states) for (−CH3) groups |
| SsssCH | MLR | Sum of E-State for all (>CH-) groups in molecule |
| SssssC | RT (2) | Sum of all (> C <) E-State value in molecule |
| TPSA | RF | Topological polar surface area (Å2) |
| VAdjEq | RF | Vertex adjacency information (equality): This is an atom count/bond count descriptor calculated as follows: − (1 − f)log2(1 − f) − f log2f where f = (n2 − m)/n2, n is the number of heavy atoms and m is the number of heavy-heavy bonds. If f is not in the open interval (0, 1), then 0 is returned. |
| vsa_hyd | BT (1) | Approximation to the sum of VDW surface areas of hydrophobic atoms (Å2) |
| vsurf_CW4 | RT (2) | Capacity factor is the ratio of the hydrophilic surface over the total molecular surface, calculated at eight different energy levels (from −0.2 to −6.0 kcal/mol) |
| vsurf_EDmin3 | RT (1) | The lowest hydrophobic energy |
| vsurf_HB4 vsurf_HB5 vsurf_HB6 | MLR, BT (1), BT (1) | H-bond donor capacity at −2.0 Kcal/mol with carbonyl oxygen probe |
| vsurf_ID7 | RT (1) | Hydrophobic integy moment (The ‘integy moment’ is defined in analogy to the dipole moment and describes the distance of the centre of mass to the barycenter of hydrophobic regions). Small integy moment indicates that the hydrophobic moieties are either close to the centre of mass or they balance at opposite ends of the molecule, so that their resulting barycentre is close to the centre of the molecule. VolSurf computes ID at eight different energy levels (from −0.2 to 1.6 Kcal/mol). |
| vsurf_IW2 | RT (2), BT (2) | Hydrophilic integy moment (see vsurf_ID7) |
| vsurf_W1 vsurf_W3 | RF, RT (1) | Hydrophilic volume |
Descriptors of RF and BTs are the top ten most important descriptors
Regression Tree Models Using CART
Several RTs were generated using a combination of molecular descriptors while cross-validation was applied (Table II). As seen in Table II, in RT (1), molecular descriptors were selected by C&RT analysis, while in RT (2), the molecular weight and in RT (3), the number of carboxylic acid groups were manually imposed as the first split descriptor using interactive C&RT routine in STATISTICA. These models were developed using the training set while the validation set remained external. The RTs resulting from these trials have been presented in Figs. 2, 3 and 4. In the regression trees, N is the number of compounds, Mu is the average and Var is the variance of LogBE% in each node. The molecular descriptors employed in the trees have been explained in Table I. Table III provides the statistical parameters of the regression trees. Observed versus calculated LogBE% plots are in supplementary material II (Figures S3 and S5) with training and validation sets identified in the plots.
Table II.
Description of the Regression Trees
| Model no | Manually incorporated variables |
|---|---|
| RT (1) | None |
| RT (2) | Molecular weight |
| RT (3) | Carboxylic acid group |
Fig. 2.
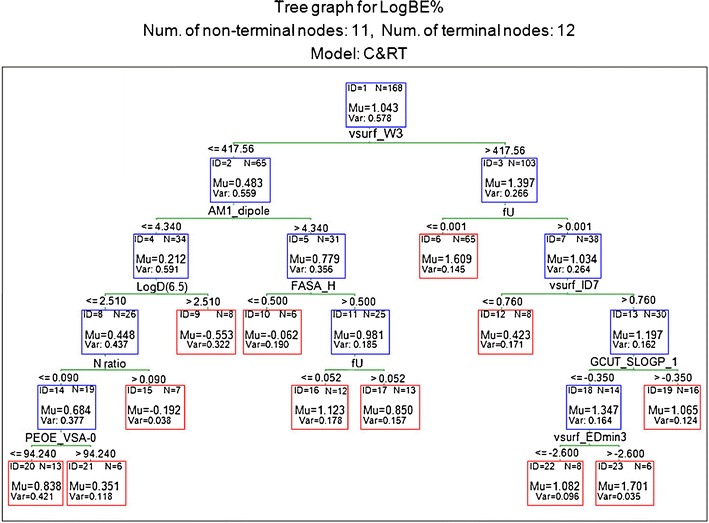
RT (1) developed using the training set with the descriptors selected by C&RT
Fig. 3.
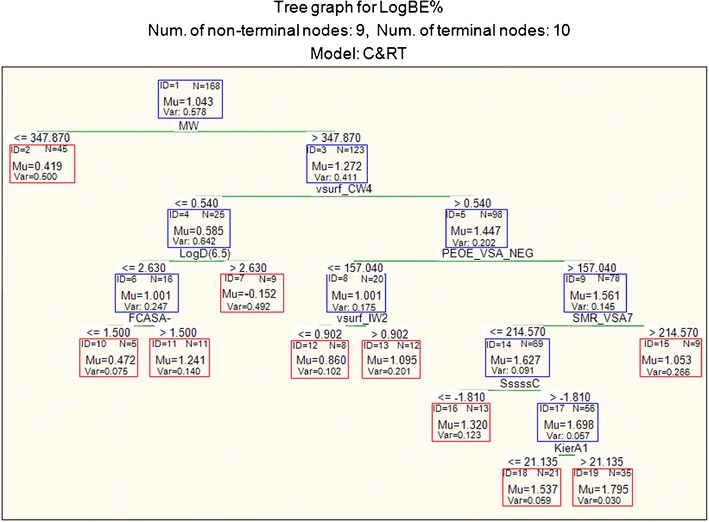
RT (2) developed using interactive C&RT analysis using molecular weight as the first descriptor
Fig. 4.
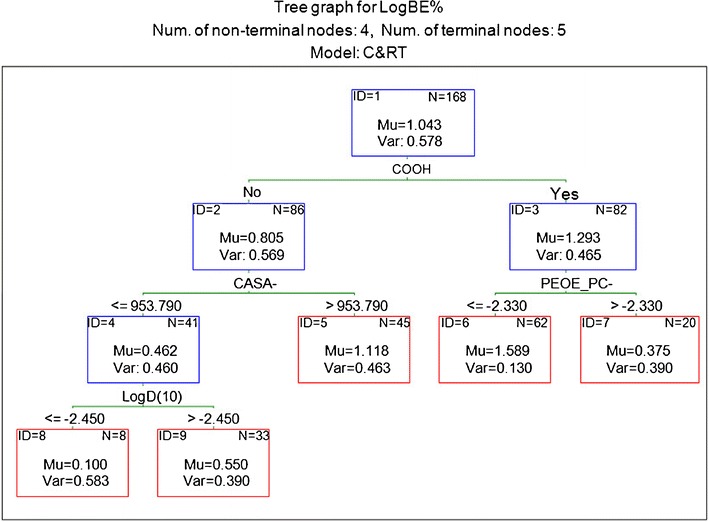
RT (3) using the number of carboxyl groups (COOH) as the first descriptor
Table III.
Statistical Parameters of the Models for Training and Test Sets
| Model | Group | Risk estimate | Standard error |
|---|---|---|---|
| RT (1) | Train | 0.112 | 0.040 |
| Validation | 0.583 | 0.116 | |
| RT (2) | Train | 0.229 | 0.034 |
| Validation | 0.348 | 0.081 | |
| RT (3) | Train | 0.323 | 0.050 |
| Validation | 0.349 | 0.075 | |
| BT (1) | Train | 0.079 | 0.007 |
| Validation | 0.328 | 0.103 | |
| BT (2) | Train | 0.078 | 0.007 |
| Validation | 0.329 | 0.107 | |
| RF | Train | 0.262 | 0.047 |
| Validation | 0.311 | 0.076 |
RT is regression tree, BT is boosted trees and RF is random forest model
According to RT (1), biliary excretion is much higher for compounds with large hydrophilic volume (vsurf_W3), especially if they are ionised with fU ≤ 0.001 (negligible unionised fractions at pH 7.4). Within the hydrophilic drugs of higher fU values (node 7), those with higher separation of lipophilic interaction sites from the centre of mass (vsurf_ID7 > 0.760) have higher biliary excretion. Surfactant molecules and glucuronide conjugates are examples of such molecules with high VolSurf integy moment (vsurf_ID7) and high biliary excretion. This branch follows to partition the molecules further according to GCUT_SLOGP_1 with compounds of lower hydrophobicity (node 18), and large hydrophobic interaction energy minima (vsurf_EDmin3 > −2.60) showing high biliary excretion (node 23). According to RT (1), the less hydrophilic drugs with vsurf_W3 values below 417.56 can be excreted heavily through the bile if they are highly dipolar (AM1-dipole >4.336) with high ratio of lipophilic to total surface area (FASA_H >0.50), especially if they are predominantly in the ionised form at pH 7.4 (fU ≤ 0.052). On the other hand, compounds with low dipole moment have low biliary excretion specially if they are lipophilic with LogD(6.5) >2.51 (node 9) or otherwise if they contain a high ratio of nitrogen atoms in the molecular structure (node 15). N ratio is low for larger alkaloids such as morphine or non-basic compounds, such as estrone 3-sulphate, which will have moderate biliary excretion especially if they are hydrophilic (PEOE_VSA-0 ≤ 94.24).
RT (2) was a result of molecular weight being employed in the first split using the interactive C&RT analysis in STATISTICA (Fig. 3). The statistically selected molecular weight threshold was 347.9 Da, with the compounds below this weight showing lower LogBE% values than the larger compounds. The tree shows that large (MW > 347.9) hydrophilic compounds (vsurf_CW4 >0.540) have higher biliary excretion, particularly those with large total negative van der Waals surface area (PEOE_VSA_NEG) and low surface area corresponding to highly polarisable groups (SMR_VSA7), especially if they are highly branched (SssssC > −1.812). Within this group of compounds, larger molecules with KierA1 >21.135 will have even higher biliary excretion. Other parameters of RT (2) indicate that high hydrophilic integy moment (vsurf_IW2) (node 13) and fractional negative charge weighted surface area (FCASA−) (node 11) would result in high LogBE% value.
Recent studies by Yang et al. (19) show that presence of carboxylic acid group(s) may indicate a trend towards increased biliary excretion. Therefore, the impact of presence of carboxylic acid group was examined using the interactive C&RT analysis with COOH used as the first partitioning molecular descriptor (Fig. 4). According to RT (3), compounds containing at least one carboxylic acid group have higher biliary excretion levels. Furthermore, RT (3) indicated that compounds with lower total negative partial charge (PEOE_PC−) have much higher biliary excretion (node 6). These are large hydrophilic compounds with many negatively charged atoms. Non-acidic compounds in node 2 will have high biliary excretion if the negative-charge weighted surface area for these molecules is high (node 5). CASA− has an element of size as well as indicating the presence of negatively charged groups such as sulphates.
Boosted Trees
Boosted tree module computes a sequence of simple trees, where each successive tree is built for the prediction of the residuals of the preceding trees. The analysis using various combination of model parameters resulted in two best models selected based on the error level for the internal test set (Table III). In models BT (1) and BT (2), the optimal numbers of trees were 145 and 147, with the learning rate of 0.10 and subsample proportions of 0.55 and 0.60, respectively.
It is possible to elucidate the influential descriptors in boosted trees analysis using variable importance calculation. Variable importance in STATISTICA is calculated as the relative (scaled) average value of the predictor statistic over all trees and nodes; hence these values reflect on the strength of the relationship between the predictors and the dependent variable of interest, over the successive boosting steps (STATISTICA help file, 2009). Included in Table I are the top ten most important molecular descriptors of BT (1) and BT (2) models. Some of the descriptors used by BT models are those already observed in RT and MLR models. For example, LogD (6.5) is present in two RT models and it is amongst the top ten most significant descriptors of both BT models. Other descriptors selected by these models are topological/size descriptors (KierA3, Kier2, Kier3) and other lipophilicity descriptors such as LogD at different pH values and vsurf descriptors. Table III shows the statistical significance of these models. Graphs of average squared error against number of trees for training and cross-validated test sets (Figures S6 and S7 for BT (1) and BT (2)) and plots of observed versus calculated LogBE% using BT (1) and BT (2) (Figures S8 and S9) can be found in supplementary material II.
Random Forest
In RF, the number of trees specifies the number of simple regression trees to be computed in successive forest building steps. The model development used the default values of the software with the number of trees set at 100. The graph of average squared error against number of trees for training and cross-validated test sets indicates that the test and training set errors reach a plateau at around 10–15 trees (see supplementary material II, Figure S10). The best model was achieved with a subsample proportion of 0.60, random test data proportion of 0.2 and number of trees of 100. Table I includes a description of the ten most significant descriptors employed in this model. Table III gives a summary of the statistical parameters of the RF model. Observed versus calculated LogBE% using the RF model can be found in supplementary material II (Figures S11).
Validation of the Models
All models were validated by the same external validation set which had been set aside and not used at any stage of model development. Table IV shows the prediction accuracy of the QSAR models using external validation in terms of the mean absolute error and the number of outliers. In addition, an average estimate of log BE% using all regression trees (RT (1)–RT (3)) was calculated and compared with the observed values to investigate any possible improvements in prediction accuracy. Table IV gives the performance of this estimation method (consensus RTs).
Table IV.
Summary of the Prediction Accuracy of the QSAR Models and the Number of Outliers with Absolute Error Above 0.6
| Model | MAE for training set | MAE for validation set | Outliers |
|---|---|---|---|
| MLR | 0.377 | 0.483 | 11 |
| RT (1) | 0.304 | 0.373 | 6 |
| RT (2) | 0.345 | 0.451 | 10 |
| RT (3) | 0.424 | 0.468 | 12 |
| Consensus RTs | 0.319 | 0.383 | 7 |
| BT (1) | 0.229 | 0.412 | 8 |
| BT (2) | 0.226 | 0.417 | 7 |
| RF | 0.403 | 0.496 | 14 |
DISCUSSION
Biliary excretion can play a significant role in the elimination of drugs, and, therefore, its prediction is an important target in drug discovery. In the pharmaceutical industry, drug candidates are routinely tested in animal studies to measure the extent of biliary excretion and propensity of enterohepatic cycling, which have significant roles in the pharmacokinetics of a drug. In drug discovery, a reliable, user friendly and low-cost model based on computer-generated molecular properties can reduce the number of high-cost animal (mainly rat) studies. This investigation aimed to elucidate how biliary excretion of compounds is controlled by their molecular structure, and to develop predictive models based on the molecular structure. Linear regression analysis, regression trees and two ensemble methods, boosted trees and random forest, were used for the QSAR model development.
Comparison of the Models
Linear regression equation is one of the simplest and the most common QSAR techniques. This method has the benefit of easy interpretation and it can provide mechanistic insight into the process under investigation. However, it has been argued that many biological processes have more complex relationships with the molecular attributes of the compounds and hence linear regression models may fail to capture these (25). RT offers a suitable alternative to MLR method with the advantage of being flexibly non-linear while retaining the interpretability (26). Ensemble methods such as random forest (22) provide consensus predictions which may have improved accuracy. But this is often accompanied by a loss of interpretability, as the ensemble of many models is often used as a ‘black box’ prediction tool. In this investigation, STATISTICA variable importance analysis was used to find the most significant molecular descriptors in the boosted trees and random forest models.
According to Table IV, the most predictive model with the lowest estimation error for the external validation set is RT (1) followed by BT (1) and BT (2) and then RT (2). In other words, increasing the complexity of the models by allowing non-linear relationships and an ensemble of such models has been able to improve the prediction accuracy in comparison with a simple linear regression model (MLR). Table IV shows the number of outliers from each of the models. According to this table, RT (1) followed by BT (2) and BT (1) and then RT (2) are the best externally validated models with the lowest numbers of outliers in the validation set. The advantage of RT is the obvious simplicity and interpretability which can make it more popular with the end users in drug discovery disciplines. For example, when using the tree for a new compound, the molecular descriptors used in the tree will need to be calculated for the compound and then the terminal node (leaf) where the compound falls according to the molecular descriptor values should be identified. The average LogBE% of the terminal node (Mu) is the estimate of the tree for this compound. Despite that RT provides discrete predictions of a continuous observation which is not ideal, this is a much more straightforward procedure than using BT or RF for the estimation of BE%. These models are ensemble of many trees, and therefore the prediction has to be performed by the computer rather than manually.
An interesting observation was made as MW and COOH were not significant in MLR equation when forced into stepwise regression analysis (P > 0.05). Despite this, incorporation of these two parameters was statistically significant in CART analysis resulting in RT (2) and RT (3). This indicates the non-linear nature of the impact of these two parameters on biliary excretion. Average prediction by the three RT models was also considered and found to be of similar accuracy to RT (1) (Table IV).
In this work, the MLR model based on the training set of 168 compounds had the second poorest prediction accuracy after RF. Studies by Yang et al. (19) and Chen et al. (23) report MLR models based on training sets of 37 and 46 compounds, respectively. The proposed model by Yang et al. incorporated molecular connectivity indexes and atom-type electrotopological indexes which have also been used in this study. The model proposed by Chen et al. also incorporated similar molecular descriptors to our study, with the addition of Abraham descriptors representing polarisability and hydrogen bond acceptor capacity. Although we have not used Abraham’s descriptors, there are other molecular descriptors in our set of 386 descriptors that measure the same properties. Examples are the number of hydrogen bonding acceptor atoms and atomic charge on the most negatively charged atom in the molecule which may represent hydrogen bond acceptor ability (27) and molar refractivity descriptors which may indicate molecular polarisability (28).
In another study, Luo et al. (17) used 50 proprietary compounds from Bristol-Myers Squibb Co. for model development. They also developed a multiple linear regression model, but in addition to more common molecular descriptors, they employed free energy of aqueous solvation calculated from a self-consistent reaction field method. In analysing this model, Gandhi and Morris (18) found that the model failed to generalise further to the new set of compounds and specifically free energy of aqueous solvation was not statistically significant. They argued that a complex process such as hepatobiliary excretion cannot be captured by simple physicochemical properties when examining chemically dissimilar compounds. Indeed, such extrapolations to external compounds will fail when the compounds are outside the domain of applicability of the QSAR models. Incorporation of a larger dataset in this work may provide the opportunity for capturing an extended chemical space. This will be discussed further when analysing the outliers in the next two sections.
Structural Features of Compounds for Biliary Excretion
Table I gives a brief description of the significant molecular descriptors used in the models. For the sake of this discussion, the descriptors in this work can be classified roughly into five categories as follows: lipophilicity, ionisation, molecular size and topological and constitutional descriptors.
It can be seen in Table I that lipophilicity descriptors such as log D at different pH levels and surface area of hydrophilic molecules (SlogP_VSA0) are present in all models. In all interpretable models (except for the linear regression equation), lipophilicity descriptors show a negative effect on the biliary excretion of compounds. This may relate to the fact that highly lipophilic compounds are known to be highly extracted and metabolised in the liver (29) rather than being excreted unchanged through bile or kidney. For example, metabolism by cytochrome P450 enzymes (30) and (UDP)-glucuronosyltransferase (31) is mainly controlled by lipophilicity and increased for more lipophilic compounds. There have been inconsistent findings in the literature regarding the effect of lipophilicity on the biliary excretion of xenobiotics. Proost et al. found no significant correlation between lipophilicity and biliary excretion of a series of bulky organic cations despite it being the predominant factor for the degree of plasma protein binding and hepatic uptake rate (29). Similar observations have been made for other compilations of biliary excretion data (19). Other studies indicate negative effect of lipophilicity on the biliary excretion within the range of compounds studied (17,32). Lipophilicity has been associated with many models of ADME properties (33). It is a well-established fact that compounds with higher logP have poor aqueous solubility and are more likely to pass through lipid bilayer of biological membranes (34). The general trend in the literature with regards to the role of lipophilicity in pharmacokinetic processes indicates that more lipophilic compounds have higher oral absorption, plasma protein binding, and volume of distribution (35–37) and are more prone to P450 metabolism (30,35). This may lead to the reduced chance of excretion through bile as the intact drug.
All models presented in this work indicate the significant role of ionisation and polarity through molecular descriptors such as COOH, fU, FCASA− and SddssS_acnt. Acids are able to ionise into anions which are substrates of several transporters (generally organic anion transporters). Compounds that carry positive as well as negative charge or partial charges can use both the ‘organic anion’ and the ‘organic cation’ transport systems (38). For example, OAT3 accepts various kinds of bulky hydrophobic anions, while OAT1 can transport relatively hydrophilic small molecules, such as nucleoside analogues (39). Besides, monocarboxylate transporters (MCT1 to MCT14) constitute a family of proton-linked plasma membrane transporters that carry molecules having one carboxylate group. MCT1 is expressed nearly all over in every tissue in the human body and also in rat and calves hepatocytes (40). MCT2 is abundant on the surface of human, rat and hamster hepatocytes (41). MCT5 and MCT8 are also known to play transporting role in rat hepatocytes (41). Studies of biliary excretion of exogenous compounds have indicated the relation between polarity and biliary excretion stating that possession of a strongly polar anionic group was important factor in appreciable biliary excretion (17,42). In all the interpretable models reported here, polarity descriptors show a positive impact on biliary excretion. Examples are the positive coefficients of dipole moment (AM1_dipole) in the linear regression equation and higher percent of compounds with lower unionised fractions at pH 7.4 (fU) in RT (1).
Molecular size is the other important factor in biliary excretion represented in the models by molecular descriptors such as kappa shape indexes, hydrophobic volumes (vsurf_W1 and vsurf_W3) and surface areas of atoms with specific charge or lipophilicity ranges (e.g. PEOE_VSA_NEG and PEOE_VSA_HYD). These molecular descriptors show positive effect on biliary excretion level in all models. This is in line with the common understanding that a molecular weight threshold may apply to biliary excretion of compounds, and that high molecular weight compounds may be predominantly excreted through bile (19,32,42). Yang et al. (19) suggested a molecular weight threshold value of 400 Da for biliary excretion of anionic drugs in rats using 164 drugs. In this study, regression tree analysis found the threshold value for molecular weight to be at 347.9 Da for biliary excretion in rat (RT (2)). Incidentally, this regression tree had the second highest prediction accuracy for the external validation set amongst the RT models. This was despite the fact that molecular weight was not the descriptor of choice by C&RT analysis.
The incorporation of some structural fragments in the models gave interesting information regarding molecular requirements for biliary excretion. Examples include SddssS_acnt and SsssCH which indicate higher biliary excretion of compounds containing sulphate groups and non-branched structure (MLR). Compounds containing carboxylic acid groups are also more likely candidates for biliary excretion according to RT (3). Up to half of compounds in our dataset contain –COOH groups (103 compounds out of 217). Sixty-five out of 103 COOH containing compounds had biliary excretion of >20%. Varma et al. (32) have analysed the interconnection between physicochemical requirements of OATP substrates and the biliary excretion rates. It was then suggested that substrate specificity of OATPs including acidity may primarily indicate the elimination through bile (32).
Analysis of the Outliers
There are a number of compounds that are outliers from majority of the models. Analysis of outliers may provide interesting information regarding the applicability of the models. Within the BE% range, it could be observed that compounds with low biliary excretion show a higher average error in general (Table V). For example, the average error by all seven models was the highest for the six compounds with the extremely low biliary excretion (BE% <0.23), followed by the compounds with 0.23 < BE% < 1.23 (−0.64 < LogBE% < 0.09). A closer inspection of the data reveals that despite the high average error for the six compounds with low biliary excretion, the estimation may still be acceptable as all these compounds have been estimated to have a BE% value <4% (average of all models) and below 0.6% by RT1 model with only one exception (benzoic acid). A hypothesis here could be that these compounds may have suitable properties for higher biliary excretion, but other routes of elimination are predominating. For example, it has been shown for benzoic acid that when clearance by the kidney is prevented, biliary excretion increases by 10% (12). Out of 217 compounds in the dataset, the predominant routes of elimination are biliary excretion for 115 compounds, renal excretion for 65 compounds and metabolism for 37 compounds. However, the outlier compounds do not belong to any single groups above in terms of the predominant routes of elimination.
Table V.
Average MAE by Nine Models for Compounds with Various BE%, logP and Molecular Weight Values
| BE% | Average MAE | n |
|---|---|---|
| ≤0.23 | 1.12 | 6 |
| 0.23–1.23 | 0.50 | 26 |
| >1.23 | 0.30 | 176 |
| MW (Da) | ||
| >280 | 0.31 | 173 |
| ≤280 | 0.54 | 35 |
| Log P | ||
| >5.35 | 0.63 | 13 |
| ≤5.35 | 0.33 | 195 |
According to Table V, highly lipophilic compounds (log P > 5.35) and low molecular weight compounds (MW ≤ 280) also show higher error rates, and this may need to be considered when using the models for the prediction of external compounds.
Table VI gives a list of the compounds that are outliers in six or seven models out of the seven models proposed here. In addition, there are four compounds which were outliers in four or five models but had exceptionally high average error from the seven models. These compounds were part of the training or validation sets, but none were omitted from average error calculations.
Table VI.
Outlier Compounds in Training or Validation Sets with Absolute Error of >0.6 in More Than Five Out of Seven Models and Their BE% Values
| Outliers | BE% | LogBE% | Over or under prediction | Models with error | MW |
|---|---|---|---|---|---|
| Benzoic acid | 0.09 | −1.07 | Over-predicted except for BT models | 4 | 122 |
| EMDP | 0.20 | −0.69 | Over-predicted | 6 | 263 |
| Fosmidomycin | 0.10 | −1.00 | Over-predicted | 7 | 183 |
| Nelfinavir | 0.05 | −1.32 | Over-predicted | 5 | 567 |
| EDDP | 36.31 | 1.56 | Under-predicted | 6 | 277 |
| PAEB | 31.62 | 1.50 | Under-predicted | 7 | 222 |
| Tolrestat | 53.70 | 1.73 | Under-predicted | 6 | 357 |
The outliers in Table VI have been over- or under-predicted by the models. One compound in the table has shown underestimation by some and overestimation by other models; biliary excretion of benzoic acid was overestimated by all models except for BT (1) and BT (2). It can be seen in Table VI that fosmidomycin, nelfinavir and 2-ethyl-5-methyl-3,3-diphenyl-1-pyrroline (EMDP) are over-predicted by five or more models. Benzoic acid is rapidly cleared by the kidney, so it may not have enough time to pass into the bile (12). Abou-El-Makarem and his colleagues examined this possibility by tying up the renal pedicles in rats, so that clearance by the kidney was prevented, and the results indicated that when clearance by the kidney is prevented, biliary excretion increased by 10% (12). Fosmidomycin has a short half-life of 1.7 h and is rapidly cleared by the kidneys (43). It is a small molecular weight polar agent which may not be cleared in high quantities through bile according to the molecular weight threshold hypothesis. Despite the use of molecular size descriptors, this compound still appeared to be overestimated by all seven models, even using RT (2) which has employed MW for the first branching. The problem with RT (2) in relation to this compound is that although this compound falls into node 2 along with 44 other low molecular weight compounds, this node has an average LogBE% of 0.42 which is much lower than node 3 with an average LogBE% of 1.27 but not low enough for this compound. Likewise, other models have indicated low biliary excretion of small-sized compounds, but somehow, estimation is higher than what is actually observed.
Nelfinavir has a half-life of 3.5 to 5 h and is eliminated via metabolism by the cytochrome P450 enzyme system (44). This is a highly lipophilic compound which is poorly excreted through bile, and is predicted as such by the models (predicted BE% below 2% using all models except for RT (3) and RF which predict 13 and 7.6%, respectively).
EMDP is a major metabolite of methadone which has been over-predicted by most models despite a very low biliary excretion. As with nelfinavir, the predicted BE% for this compound by most models is quite low at <4% (MLR is an exception) and the selected model, RT (1), predicts a biliary excretion value of ∼0.3%. Despite this, in comparison with the extremely low observed value of 0.05%, the predicted values are much higher, leading to a numerically large average error, even though qualitatively, the predicted biliary excretion may be reasonably low.
EDDP, PAEB (procaine amide ethobromide) and tolrestat are the under-predicted compounds. All these compounds have high BE% values at 36, 32 and 54%. This is despite the relatively low molecular weights of EDDP and PAEB which are below the defined MW threshold of 347 Da for biliary excretion. The exact mechanism of high biliary excretion of these compounds warrants further investigation to explore the reasons behind such high biliary excretion despite the low molecular weight.
Tolrestat has a relatively high molecular weight suitable for biliary excretion and a COOH group making it a suitable substrate for OATPs (32). Despite this, the hydrophilic volume calculated by the VolSurf descriptor vsurf-W3 is not high enough to put this compound in node 3 rather than node 2 of RT (1) model. In RT (2), the compound falls into node 16, which is due to the lack of non-aromatic branched structure which would place it in node 17 with a higher predicted BE%. Likewise, in RT (3), this compound fails to be placed in node 7 and falls in node 6 instead due to the low total negative charge (> −2.33) as a result of the low number of negatively charged atoms. This indicates a shortcoming in the abovementioned models which lack suitable parameters that can capture the relative polarity in relation to the molecular size.
CONCLUSION
This investigation focused on the development of computational models for a cost-effective estimation of biliary excretion of compounds. This was made possible through the application of quantitative structure-activity relationships where molecular properties (descriptors) of a large dataset of compounds were related to the percentage of dose excreted intact via the bile through the use of statistical techniques. Some of the statistical techniques led to very promising results as evaluated by the prediction accuracy for the external validation set. The QSAR models also identified the important molecular properties (descriptors) that have the main influence on biliary excretion of compounds. The selected models were the regression tree (CART) model, RT (1), followed by boosted trees models BT (1) and BT (2). Regression trees also have the advantage of being simple, interpretable and user-friendly. The models generally indicated that larger, relatively hydrophilic molecules containing a carboxylic acid group are more prone to biliary excretion. For example, in the selected model, RT (1), compounds with increased hydrophilic volume and acidic dissociation have high biliary excretion. The significance of acidity and molecular size were further confirmed through interactive regression trees and a statistically validated MW threshold for effective biliary excretion was established. Detailed analysis of the error levels and outliers indicated that the models work best for larger compounds (MW > 280 Da) and are less accurate for extremely lipophilic compounds (log P > 5.35).
Electronic supplementary material
(XLSX 36 kb)
(DOCX 173 kb)
References
- 1.Rosenbaum SE. Basic pharmacokinetics and pharmacodynamics, an integrated textbook and computer simulations. 1. Hoboken: John Wiley and Sons; 2011. [Google Scholar]
- 2.Rollins DE, Klaassen CD. Biliary excretion of drugs in man. J Clin Pharmacokinet. 1979;4:368–379. doi: 10.2165/00003088-197904050-00003. [DOI] [PubMed] [Google Scholar]
- 3.Kullak-Ublick GA, Stieger B, Hagenbuch B, Meier PJ. Hepatic transport of bile salts. Semin Liver Dis. 2000;20:273–292. doi: 10.1055/s-2000-9426. [DOI] [PubMed] [Google Scholar]
- 4.Van Montfoort JE, Hagenbuch B, Groothuis GM, Koepsell H, Meier PJ, Meijer DK. Drug uptake systems in liver and kidney. Curr Drug Metab. 2003;4:185–211. doi: 10.2174/1389200033489460. [DOI] [PubMed] [Google Scholar]
- 5.Leabman MK, Huang CC, DeYoung J, Carlson EJ, Taylor TR, De la Cruz M, et al. Natural variation in human membrane transporter genes reveals evolutionary and functional constraints. Proc Natl Acad Sci U S A. 2003;100:5896–5901. doi: 10.1073/pnas.0730857100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nies AT, Koepsell H, Winter S, Burk O, Klein K, Kerb R, et al. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology. 2009;50:1227–1240. doi: 10.1002/hep.23103. [DOI] [PubMed] [Google Scholar]
- 7.Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function and regulation. Physiol Rev. 2003;83:633–671. doi: 10.1152/physrev.00027.2002. [DOI] [PubMed] [Google Scholar]
- 8.Morgan RE, Trauner M, van Staden CJ, Lee PH, Ramachandran B, Eschenberg M, et al. Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol Sci. 2010;118:485–500. doi: 10.1093/toxsci/kfq269. [DOI] [PubMed] [Google Scholar]
- 9.Schinkel AH, Mayer U, Wagenaar E, Mol CA, Deemter LV, Smit JJ, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci U S A. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merino G, Jonker JW, Wagenaar E, van Herwaarden AE, Schinkel AH. The breast cancer resistance protein (BCRP/ABCG2) affects pharmacokinetics, hepatobiliary excretion and milk secretion of the antibiotic nitrofurantoin. Mol Pharmacol. 2005;67:1758–1764. doi: 10.1124/mol.104.010439. [DOI] [PubMed] [Google Scholar]
- 11.Hirom PC, Millburn P, Smith RL, Williams RT. Species variations in the threshold molecular-weight factor for the biliary excretion of organic anions. Biochem J. 1972;129:1071–1077. doi: 10.1042/bj1291071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abou-El-Makarem MM, Millburn P, Smith RL, Williams RT. Biliary excretion in foreign compounds. Species difference in biliary excretion. Biochem J. 1967;105:1269–1274. doi: 10.1042/bj1051289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crosignani A. Clinical pharmacokinetics of therapeutic bile acids. J Clin Pharmacokinet. 1996;30:333–358. doi: 10.2165/00003088-199630050-00002. [DOI] [PubMed] [Google Scholar]
- 14.Neef C, Keulemans KT, Meijer DK. Hepatic uptake and biliary excretion of organic cations–I. Characterization of three new model compounds. Biochem Pharmacol. 1984;33:3977–3990. doi: 10.1016/0006-2952(84)90011-X. [DOI] [PubMed] [Google Scholar]
- 15.Feitsma KG. Unequal disposition of enantiomers of the organic cation oxyphenonium in the rat isolated perfused liver. J Pharm Pharmacol. 1989;41:27–31. doi: 10.1111/j.2042-7158.1989.tb06323.x. [DOI] [PubMed] [Google Scholar]
- 16.Ghafourian T, Barzegar-Jalali M, Dastmalchi S, Khavari-Khorasani T, Hakimiha N, Nokhodchi A. QSPR models for the prediction of apparent volume of distribution. Int J Pharm. 2006;319:82–97. doi: 10.1016/j.ijpharm.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 17.Luo G, Johnson S, Hsueh M, Zheng J, Hong C, Xin B, et al. In silico prediction of biliary excretion of drugs in rats based on physicochemical properties. Drug Metab Dispos. 2010;38:422–430. doi: 10.1124/dmd.108.026260. [DOI] [PubMed] [Google Scholar]
- 18.Gandhi YA, Morris ME. Re-evaluation of a quantitative structure pharmacokinetic model for biliary excretion in rats. Drug Metab Dispos. 2012;40:1259–1262. doi: 10.1124/dmd.112.044842. [DOI] [PubMed] [Google Scholar]
- 19.Yang X, Gandhi YA, Duignan DB, Morris ME. Prediction of biliary excretion in rats and humans using molecular weight and quantitative structure–pharmacokinetic relationships. AAPS. 2009;11:511–525. doi: 10.1208/s12248-009-9124-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breiman L, Friedman JH, Olshen RA, Stone CJ. Classification and regression trees. Monterey: Wadsworth & Brooks/Cole Advanced Books & Software; 1984. [Google Scholar]
- 21.Lewicki P, Hill S. Statistics, methods and applications, a comprehensive reference for science, industry and data mining. 1. USA: StatSoft Inc; 2006. [Google Scholar]
- 22.Breiman L. Random forests. Mach Learn. 2001;45:5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 23.Chen Y, Cameron K, Guzman-Perez A, Perry D, Li D, Gao H. Structure–pharmacokinetic relationship of in vivo rat biliary excretion. Biopharm Drug Dispos. 2010;31:82–90. doi: 10.1002/bdd.692. [DOI] [PubMed] [Google Scholar]
- 24.Zamek-Gliszczynski MJ, Hoffmaster KA, Humphreys JE, Tian X, Nezasa K, Brouwer KLR. Differential involvement of Mrp2 (Abcc2) and Bcrp (Abcg2) in biliary excretion of 4-methylumbelliferyl glucuronide and sulphate in the rat. J Pharmacol Exp Ther. 2006;319:459–467. doi: 10.1124/jpet.106.101840. [DOI] [PubMed] [Google Scholar]
- 25.Guha R, Jurs PC. Development of linear, ensemble, and nonlinear models for the prediction and interpretation of the biological activity of a set of PDGFR inhibitors. J Chem Inf Comput Sci. 2004;44:2179–2189. doi: 10.1021/ci049849f. [DOI] [PubMed] [Google Scholar]
- 26.De’ath G, Fabricius KE. Classification and regression trees: a powerful yet simple technique for ecological data analysis. Ecology. 2000;81:3178–3192. doi: 10.1890/0012-9658(2000)081[3178:CARTAP]2.0.CO;2. [DOI] [Google Scholar]
- 27.Dearden JC, Ghafourian T. Hydrogen bonding parameters for QSAR: comparison of indicator variables, hydrogen bond counts, molecular orbital and other parameters. J Chem Inf Comput Sci. 1999;39:231–235. doi: 10.1021/ci980065+. [DOI] [Google Scholar]
- 28.Verma RP, Hansch C. A comparison between two polarizability parameters in chemical–biological interactions. Bioorg Med Chem. 2005;13:2355–2372. doi: 10.1016/j.bmc.2005.01.051. [DOI] [PubMed] [Google Scholar]
- 29.Proost JH, Roggeveld J, Wierda JM, Meijer DK. Relationship between chemical structure and physicochemical properties of series of bulky organic cations and their hepatic uptake and biliary excretion rates. J Pharmacol Exp Ther. 1997;282:715–726. [PubMed] [Google Scholar]
- 30.Lewis DF, Ito Y. Human CYPs involved in drug metabolism: structures, substrates and binding affinities. Expert Opin Drug Metab Toxicol. 2010;6:661–674. doi: 10.1517/17425251003674380. [DOI] [PubMed] [Google Scholar]
- 31.Smith PA, Sorich MJ, McKinnon RA, Miners JO. In silico insights: chemical and structural characteristics associated with uridine diphosphate glucuronosyltransferase substrate selectivity. Clin Exp Pharmacol Physiol. 2003;30:836–840. doi: 10.1046/j.1440-1681.2003.03923.x. [DOI] [PubMed] [Google Scholar]
- 32.Varma MV, Chang G, Lai Y, Feng B, El-Kattan AF, Litchfield J, et al. Physicochemical property space of hepatobiliary transport and computational models for predicting rat biliary excretion. Drug Metab Dispos. 2012;40:1527–1537. doi: 10.1124/dmd.112.044628. [DOI] [PubMed] [Google Scholar]
- 33.Hansch C, Leo A, Mekapati SB, Kurup A. QSAR and ADME. Bioorg Med Chem. 2004;12:3391–3400. doi: 10.1016/j.bmc.2003.11.037. [DOI] [PubMed] [Google Scholar]
- 34.Kerns EH, Di L. Drug-like properties: concepts, structure, design and methods. 1. London: Elsevier; 2008. [Google Scholar]
- 35.van de Waterbeemd H, Smith DA, Jones BC. Lipophilicity in PK design: methyl, ethyl, futile. J Comput Aided Mol Des. 2001;15:273–286. doi: 10.1023/A:1008192010023. [DOI] [PubMed] [Google Scholar]
- 36.Obach RS, Lombardo F, Waters NJ. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 670 drug compounds. Drug Metab Dispos. 2008;36:1385–1405. doi: 10.1124/dmd.108.020479. [DOI] [PubMed] [Google Scholar]
- 37.Newby D, Freitas AA, Ghafourian T. Coping with unbalanced class datasets in oral absorption models. J Chem Inf Model. 2013;53:461–474. doi: 10.1021/ci300348u. [DOI] [PubMed] [Google Scholar]
- 38.Koepsell H, Gorboulev, Popp C, van Montfoort JE, Meier PJ, Arndt P, et al. Hepatobiliary transport from bench to beside. London: Kluwer; 2001. Organic cation transporters in the sinusoidal membrane of hepatocytes; pp. 3–15. [Google Scholar]
- 39.Maeda K, Shitara Y, Horie T, Sugiyama Y. Web-based database as a tool to examine drug-drug interactions involving transporters. In: Pang SK, Rodrigues DA, Raimund MP, editors. Enzyme and transporter-based drug-drug interactions. Progress and future challenges. London: Springer; 2010. pp. 387–414. [Google Scholar]
- 40.Kirat D, Inoue H, Iwano H, Yokota H, Taniyama H, Kato S. Monocarboxylate transporter 1 (MCT1) in the liver of pre-ruminant and adult bovines. Vet J. 2007;173:124–130. doi: 10.1016/j.tvjl.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 41.Halestrap AP, Meredith D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004;447:619–628. doi: 10.1007/s00424-003-1067-2. [DOI] [PubMed] [Google Scholar]
- 42.Millburn R, Smith RL, Williams RT. Biliary excretion of foreign compounds. Biochem J. 1967;105:1275–1281. doi: 10.1042/bj1051275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murakawa T, Sakamoto H, Fukada S, Konishi T, Nishida M. Pharmacokinetics of fosmidomycin: a new phosphonic acid antibiotic. Antimicrob Agents Chemother. 1982;21:224–230. doi: 10.1128/AAC.21.2.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bardsleey-Elliot A, Plosker GL. Nelfinavir an update on its use in HIV infection. Drugs. 2000;59:581–620. doi: 10.2165/00003495-200059030-00014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX 36 kb)
(DOCX 173 kb)