

Abstract
BACKGROUND
Genetic mutations play crucial role in the etiology of cryptogenic infantile spasms but the cause is still unknown in a significant proportion of patients. Whole exome sequencing technology shows great promise in identifying genetic causes of infantile spasms.
METHODS
We performed whole exome sequencing and 2-deoxy-2-(18F)fluoro-D-glucose positron emission tomography scan of a male child with infantile spasms. Exome sequencing was also performed in parents to identify any de novo mutations.
RESULTS
The positron emission tomography scan showed a pattern of bilateral symmetric temporal lobe glucose hypometabolism. A total of 8171 non-synonymous variants were identified in the child. Despite the large number of nonsynonymous variants, there was only a single de novo missense mutation in SCN2A in the child (NCBI hg19 assembly, position: Chr2:166234116, K1422E). Subsequent Sanger sequencing confirmed the de novo status of this variant. This mutation has never been reported in 6500 individuals of the exome variant server database. Similarly, this variant is not reported in online mendelian inheritance in man database or human gene mutation database. It has previously been shown that SCN2A mutations are associated with hippocampal hyperexcitability. Therefore, we speculate that IS and bitemporal hypometabolism in our patient might have been caused by hippocampal hyperexcitability due to SCN2A mutation.
CONCLUSIONS
The simultaneous presence of an SCN2A mutation and bitemporal hypometabolism in this patient with infantile spasms suggests a plausible hippocampal origin. However, additional mechanistic and clinical studies are required to validate this link.
Keywords: Infantile Spasms, Exome sequencing, Positron Emission Tomography, Genetics, Ion channel gene defects
Introduction
Infantile spasms (IS) are epileptic seizures characterized by brief muscle extension, flexion, or mixed flexion-extension jerks involving the neck, trunk, or extremities and beginning between 3 and 8 months of age [1] with an incidence of 0.25 to 0.4 per 1000 live births [2]. Both genetic and environmental causes have been implicated in IS [3]. In a recent study (UKISS) of 207 infants with IS, only 127 (61%) cases had an identified underlying etiology, the most common of which were hypoxic ischemic encephalopathy (21), chromosomal abnormalities (16), brain malformations (16), stroke (16), and tuberous sclerosis complex (TSC) (15) [4]. The remaining 32 etiologies were all individually uncommon [4]. Yet, in a study of familial epilepsy in Sweden, IS had the highest sibling recurrence risk ratio of 10.45, suggesting a significant role of genetic mechanisms in cryptogenic IS [5].
Several genetic causes of IS have been identified and summarized [6]. Despite this progress, the cause is still unknown in a significant proportion of patients. Recent advances in next generation sequencing technologies show great promise in identifying additional causes of IS. However, the massive datasets generated by these technologies introduce new challenges in data analysis. In addition, pathogenic mechanisms of mutations of IS (such as abnormalities in specific biochemical pathways, cell types, anatomical structures etc) are yet to be defined adequately. Thus, integration of additional information from other sources such as neuroimaging may shed light into the identification of the underlying etiologies and pathogenic mechanisms. For example, 2-deoxy-2-(18F)fluoro-D-glucose positron emission tomography (FDG-PET) scanning of glucose metabolism can provide important clues and insights into the pathogenic mechanisms of IS. FDG-PET scans show four different glucose metabolic patterns (focal, multifocal, diffuse and bitemporal cortical hypometabolism) in cryptogenic IS. The focal and multifocal patterns indicate underlying cortical dysplasias which may or may not be apparent on MRI [7, 8]. The diffuse pattern suggests abnormalities involving global brain processes such as synaptic physiology or neuronal metabolism. Interestingly, the bitemporal hypometabolism pattern appears to be a biomarker of autism in this population and is associated with a rather specific phenotype [9] and could localize the functional abnormality to medial or lateral temporal lobes. Therefore, FDG-PET scan could be used as an imaging endophenotype.
In the present case report, we performed whole exome sequencing in an IS patient who had bitemporal glucose hypometabolism on FDG-PET. We reasoned that performing exome sequencing in an IS patient with a specific imaging endophenotype (such as this patient) may facilitate the identification of new genetic causes and may help in suggesting the underlying mechanism of IS.
Case Report
We report the findings on whole exome sequencing and 2-deoxy-2-(18F)fluoro-D-glucose positron emission tomography (FDG-PET) scan of a male child with IS. The parents are completely normal without history of seizures or epilepsy. The child appeared normal until age 4 months when poor eye contact and weight-bearing were noted. Between 4 and 6 months, he developed episodes of left-sided clonus and mandibular shivering. At 12 months of age, after vaccination, he developed fever and flexor spasms. He was treated unsuccessfully with levetiracetam, high dose prednisolone, vigabatrin and ketogenic diet. ACTH for 6 weeks resolved his hypsarrythmia but spasms persisted. He also had features suggestive of autism such as repetitive movements and poor eye contact. The MRI, chromosome microarray and routine metabolic testing and extensive clinical genetic testing did not show any abnormality. The FDG-PET scan showed a pattern of bilateral symmetric temporal lobe hypometabolism (Figure 1).
Figure 1.
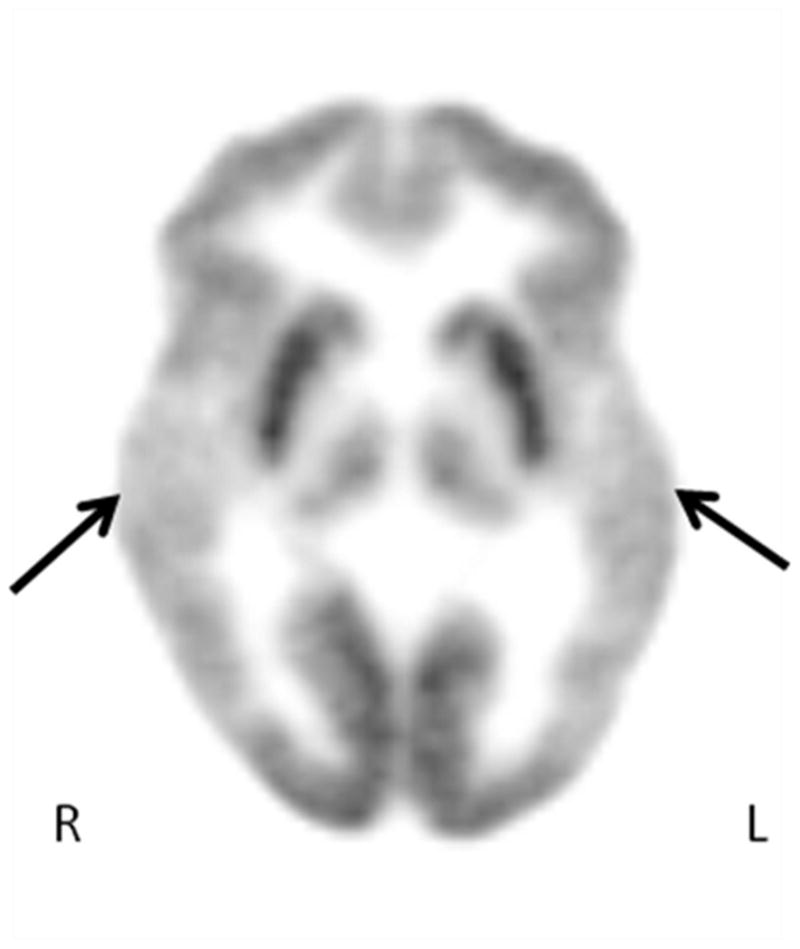
Representative axial image of FDG-PET scan showing the hypometabolism involving bilateral temporal lobes (see the arrows). The arrows correspond to both temporal lobes. Note the severe hypometabolism of both temporal lobes and sparing of other lobes.
Exome sequencing was performed for the whole family (approved by Wayne State University Institutional Review Board and informed consent obtained from the family). Briefly, Agilent V4 (51 MB) human exome enrichment kit was used to extract the exome library from five micrograms of genomic DNA. The sequencing was performed using Illumina HiSeq2000. All the raw reads were mapped to a reference human genome (hg 19, Feb 2009) with a pattern based mapper developed by DNAnexus, that is quite similar to other pattern-based mapping tools such as ELAND and MAQ (described in detail in the website: https://dnanexus.com/whitepapers/DNAnexus-whitepaper-read-mapping.pdf). Subsequently, a probabilistic model was used to identify single nucleotide variants. This method is described in detail in the following website: https://dnanexus.com/whitepapers/DNAnexus-whitepaper-nucleotide-level-variation.pdf. Variants with a read depth of less than 20 were discarded. SeattleSeq was used to annotate the identified variants (http://snp.gs.washington.edu/SeattleSeqAnnotation137/index.jsp). An in-house workflow was subsequently used to detect the unique variants in the child with normal sequence in the parents (i.e., de novo mutation).
The mean coverage across the whole exome was 80.22. A total of 8171 non-synonymous variants were identified in the child. Despite the large number of nonsynonymous variants, there was only a single de novo missense mutation in SCN2A in the child (NCBI hg19 assembly, position: Chr2:166234116, K1422E) (Figure 2). Subsequent Sanger sequencing confirmed the de novo status of this variant (Figure 3). This mutation has never been reported in 6500 individuals of the exome variant server database. Similarly, this variant is not reported in online mendelian inheritance in man (OMIM) database or human gene mutation database (HGMD).
Figure 2.
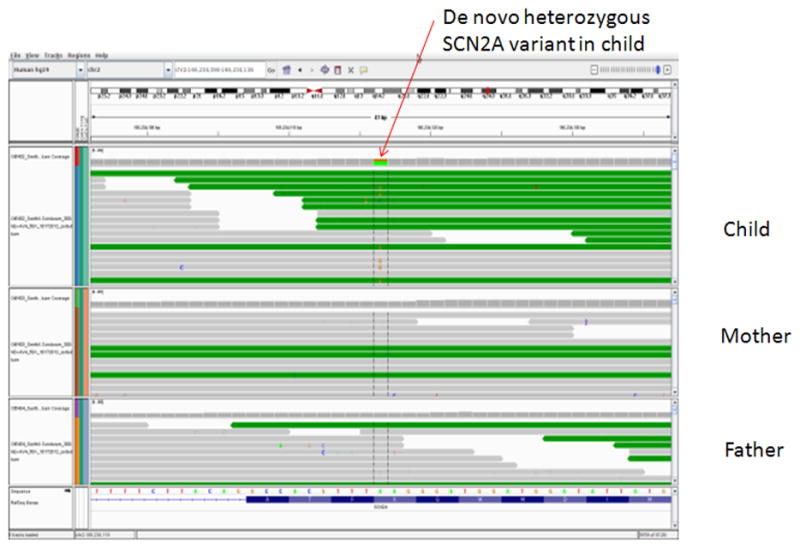
showing the de novo heterozygous mutation in SCN2A. The figure shows the genome browser view of the variant (position - Chr2:166234116, NCBI hg19 assembly) and its neighborhood in the child and parents. No SNP was identified in either parent (corresponding to the reference genotype of AA) whereas the child possessed a SNP with the genotype of AG (shown in green/orange color).
Figure 3.
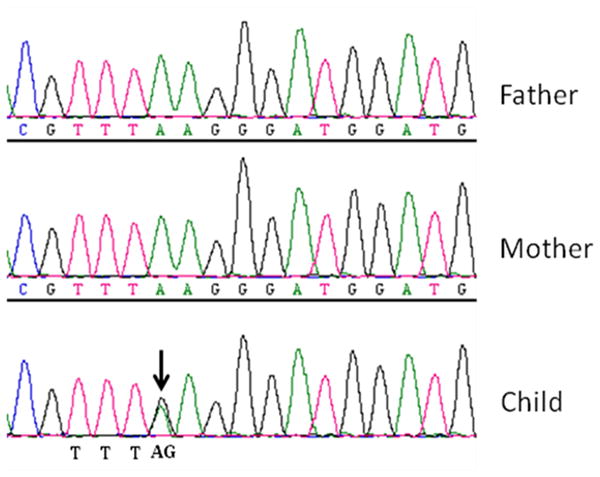
showing the validation of the heterozygous missense mutation in SCN2A using Sanger sequencing. Sanger sequencing confirmed the genotypes of the child (AG) and the parents (AA) initially identified by the nextgen sequencing.
Discussion
In this IS child with bitemporal glucose hypometabolism on PET scanning, we identified a de novo mutation in a sodium channel gene, SCN2A. A different mutation in SCN2A has so far been reported in a single patient with infantile spasms [10] but the FDG-PET scan pattern is unknown in this patient. Mutations in several other genes have been implicated in IS and they have been classified into different categories [6]. Mutations in these genes are both de novo and inherited variants, and some mutations show genetic pleiotropy and cause variable neurodevelopmental phenotypes such as autism and intellectual disability. Thus, despite significant progress, neither the general characteristics of IS mutations nor the underlying pathogenic mechanisms have been characterized fully.
An average newborn is calculated to have acquired 50 to 100 new mutations in his or her genome, resulting in approximately 0.86 de novo, nonsynonymous mutations [11]. The identification of a single de novo nonsynonymous variant in this child is consistent with this known frequency of de novo, nonsynonymous mutations [11]. Such spontaneous germline mutations can have serious phenotypic consequences when they affect functionally relevant genes. However, such de novo mutations can also be private variants of little consequence if they occur in functionally redundant genes. Thus, evaluation of the functional consequences of mutation can validate the pathogenic significance of the identified variant. While functional validation is typically performed using cellular and animal experiments, a presumptive importance of the identified variant can sometimes be inferred directly in patients using functional imaging such as FDG-PET.
The presence of bitemporal glucose hypometabolism in this patient may lead one to localize the functional abnormality to medial or lateral temporal lobes, though this needs to be further validated in future studies. In general, there are 4 PET patterns associated with cryptogenic IS: focal, multifocal, diffuse, and bitemporal hypometabolism [7]. The most common of these involves multifocal hypometabolism likely to be caused by abnormal neuronal proliferation/differentiation mechanisms typically involving more than one lobe. The unifocal cases usually have an underlying cortical dysplasia and are candidates for focal cortical resection [8]. The patient in the present report had the pattern of bilateral temporal lobe hypometabolism seen in 10–15% of cryptogenic IS patients [7]. This subgroup has a relatively consistent phenotype, characterized by low functioning autistic disorder, bitemporal epileptiform discharges and severe delay/absence of speech [9]. It is well known that SCN2A mutations are associated with hippocampal hyperexcitability [12]. Therefore, we speculate that IS and bitemporal hypometabolism in our patient might have been caused by hippocampal hyperexcitability due to SCN2A mutation. Lesional epilepsy due to dysplasias, migrational disorders, hamartomas, and low-grade tumors (such as gangliogliomas) may cause focal and unilateral temporal lobe involvement [13], but would unlikely cause symmetric bitemporal hypometabolism. Similarly, genes affecting generalized neocortical synaptic dysfunction are likely to cause diffuse cortical hypometabolism involving all lobes instead of specific bitemporal hypometabolism. In this regard, the hippocampal hyperexcitability caused by SCN2A is an attractive candidate mechanism that can result in this PET pattern. Although SCN2A has previously been found in a single patient with IS [10], its association with bitemporal hypometabolism has not been previously reported and may explain the endophenotype of bitemporal hypometabolism, which could be a PET biomarker of this IS subgroup. While the simultaneous presence of an SCN2A mutation and bitemporal hypometabolism in this patient suggests a plausible hippocampal origin (as noted above), more mechanistic and clinical studies are clearly required to validate this link.
References
- 1.Kellaway P, Hrachovy RA, Frost JD, Jr, Zion T. Precise characterization and quantification of infantile spasms. Ann Neurol. 1979;6:214–218. doi: 10.1002/ana.410060306. [DOI] [PubMed] [Google Scholar]
- 2.Trevathan E, Murphy CC, Yeargin-Allsopp M. The descriptive epidemiology of infantile spasms among Atlanta children. Epilepsia. 1999;40:748–751. doi: 10.1111/j.1528-1157.1999.tb00773.x. [DOI] [PubMed] [Google Scholar]
- 3.Riikonen R, Donner M. Incidence and aetiology of infantile spasms from 1960 to 1976: a population study in Finland. Dev Med Child Neurol. 1979;21:333–343. doi: 10.1111/j.1469-8749.1979.tb01625.x. [DOI] [PubMed] [Google Scholar]
- 4.Osborne JP, Lux AL, Edwards SW, et al. The underlying etiology of infantile spasms (West syndrome): information from the United Kingdom Infantile Spasms Study (UKISS) on contemporary causes and their classification. Epilepsia. 2010;51:2168–2174. doi: 10.1111/j.1528-1167.2010.02695.x. [DOI] [PubMed] [Google Scholar]
- 5.Hemminki K, Li X, Johansson SE, Sundquist K, Sundquist J. Familial risks for epilepsy among siblings based on hospitalizations in Sweden. Neuroepidemiology. 2006;27:67–73. doi: 10.1159/000094976. [DOI] [PubMed] [Google Scholar]
- 6.Paciorkowski AR, Thio LL, Dobyns WB. Genetic and biologic classification of infantile spasms. Pediatr Neurol. 1998;45:355–367. doi: 10.1016/j.pediatrneurol.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chugani HT, Conti JR. Etiologic classification of infantile spasms in 140 cases: role of positron emission tomography. J Child Neurol. 1996;11:44–48. doi: 10.1177/088307389601100111. [DOI] [PubMed] [Google Scholar]
- 8.Chugani HT, Shields WD, Shewmon DA, Olson DM, Phelps ME, Peacock WJ. Infantile spasms: I. PET identifies focal cortical dysgenesis in cryptogenic cases for surgical treatment. Ann Neurol. 1990;27:406–413. doi: 10.1002/ana.410270408. [DOI] [PubMed] [Google Scholar]
- 9.Chugani HT, Da Silva E, Chugani DC. Infantile spasms: III. Prognostic implications of bitemporal hypometabolism on positron emission tomography. Ann Neurol. 1996;39:643–649. doi: 10.1002/ana.410390514. [DOI] [PubMed] [Google Scholar]
- 10.Ogiwara I, Ito K, Sawaishi Y, et al. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology. 2009;73:1046–1053. doi: 10.1212/WNL.0b013e3181b9cebc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U S A. 2010;107:961–968. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kile KB, Tian N, Durand DM. Scn2a sodium channel mutation results in hyperexcitability in the hippocampus in vitro. Epilepsia. 2008;49:488–499. doi: 10.1111/j.1528-1167.2007.01413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bourgeois BF. Temporal lobe epilepsy in infants and children. Brain Dev. 1998;20:135–141. doi: 10.1016/s0387-7604(98)00010-2. [DOI] [PubMed] [Google Scholar]