

Background: Protons modify cardiac sodium channel function, potentially contributing to cardiac arrhythmia during and following ischemia.
Results: Protons binding amino acids at the pore alter cardiac sodium channel function.
Conclusion: Sodium channel pore protonation mediates proton block and destabilization of sodium channel slow inactivation.
Significance: Understanding sodium channel proton modulation is necessary to understand the pathophysiology of cardiac acidosis.
Keywords: Acidosis, Electrophysiology, Ion Channels, Ischemia, Sodium Channels, Cut-open Voltage Clamp, Proton Block, Slow Inactivation
Abstract
Protons impart isoform-specific modulation of inactivation in neuronal, skeletal muscle, and cardiac voltage-gated sodium (NaV) channels. Although the structural basis of proton block in NaV channels has been well described, the amino acid residues responsible for the changes in NaV kinetics during extracellular acidosis are as yet unknown. We expressed wild-type (WT) and two pore mutant constructs (H880Q and C373F) of the human cardiac NaV channel, NaV1.5, in Xenopus oocytes. C373F and H880Q both attenuated proton block, abolished proton modulation of use-dependent inactivation, and altered pH modulation of the steady-state and kinetic parameters of slow inactivation. Additionally, C373F significantly reduced the maximum probability of use-dependent inactivation and slow inactivation, relative to WT. H880Q also significantly reduced the maximum probability of slow inactivation and shifted the voltage dependence of activation and fast inactivation to more positive potentials, relative to WT. These data suggest that Cys-373 and His-880 in NaV1.5 are proton sensors for use-dependent and slow inactivation and have implications in isoform-specific modulation of NaV channels.
Introduction
Voltage-gated sodium (NaV)4 channels are responsible for action potential propagation in most excitable cells. Each channel is composed of a pore-forming α-subunit and one or more modulating β-subunits (1, 2). The α-subunit forms the functional pore of the channel and is composed of four homologous domains (DI–DIV) consisting of six, transmembrane helices (S1–S6) (1, 3). Helices S1–S4 form the channel's voltage-sensing domain, primarily mediated by a high concentration of positively charged arginines and lysines positioned at every third residue within each S4 helix (4). The S5 and S6 helices and the extracellular loops linking them (p-loops) combine to form the functional pore and selectivity filter of the channel (5).
Two processes regulate NaV channel availability: fast inactivation (FI) and slow inactivation (SI). FI is believed to occur through occlusion of the intracellular mouth of the pore by the intracellular DIII-DIV linker (6). This process is called FI because it occurs in the time frame of several milliseconds. In contrast, NaV channels undergo slow inactivation in response to prolonged (>0.5-s) depolarization or rapid, successive depolarizations. This process is known as SI because its onset and recovery occur in the time frame of seconds to tens of seconds (7). SI is also molecularly and pharmacologically distinct from FI (7). Intermediate, slow, and ultraslow inactivated states have been described and occur in the 0.5–1 s, 1–60 s, and >60 s time domains, respectively (8, 9). The presence of sequential inactivated states, dependent on the duration and frequency of depolarization and distinct from FI, was suggested to be a continuum of slow inactivated states (7).
The mechanism of SI is poorly understood but is thought to be a collapse of the pore mediated through long distance interactions within the channel. Several mutations at the external pore disrupt SI (8–10). Metal cations at the external pore inhibit SI (11), and structural rearrangement at the external pore occurs concurrently with SI onset (12, 13). Further, the probability that a channel will slow inactivate varies between isoforms (∼50% in NaV1.5 and ∼90% in NaV1.4), a difference that is mediated by p-loop residues of the DII S5-S6 linker (8, 14). C-type inactivation, the potassium channel equivalent of NaV channel SI, involves constriction at the selectivity filter coupled with the movement of the activation gate (15, 16). Most recently, several crystal structures of bacterial NaV channels suggest that collapse of the channel's selectivity filter contributes to slow inactivation (17, 18).
Additionally, mutagenic replacement of residues Ile-1303, Phe-1304, and Met-1305 (NaV1.4 numbering) of the DIII-DIV linker with QQQ abolishes FI but increases the probability for SI (19). Mutating the negatively charged residues adjacent to the IFM sequence alters the properties of SI (20). Further, mutations involving the cytoplasmic S4-S5 linkers as well as the S4, S5, and S6 helices have been shown to affect NaV channel SI (21–28). Capes et al. (29) showed that prolonged depolarizations that induced slow inactivation inhibited gating pore currents through the DIV voltage-sensing domain and demonstrated that movements within the NaV channel voltage-sensing domain and selectivity filter are coupled.
Protons destabilize the fast and slow inactivated states of NaV1.5 channels, the primary NaV channel isoform found in cardiac tissue. During cardiac ischemia, extracellular pH drops from pH 7.4 to as low as pH 6.0 within ∼10 min of its onset (30). Changes in NaV1.5 channel function during cardiac ischemia are believed to contribute to cardiac arrhythmia (31). Protonation of p-loop amino acids causes a reduction of single channel conductance, in skeletal muscle NaV1.4 channels, with a pKa of pH ∼6.0 and complete block at pH ∼4.0 (32, 33). The amino acids that may underlie changes in NaV1.5 channel gating during extracellular acidosis are unknown.
The goal of this study was to identify proton-sensitive residues that mediate the changes in NaV1.5 channel kinetics during extracellular acidosis. We expressed wild-type and mutant constructs of the human variant of the cardiac NaV channel, hNaV1.5, in Xenopus oocytes. We demonstrate that p-loop residues mediate proton modulation and block of NaV1.5 channels and contribute to NaV1.5 channel SI stability.
MATERIALS AND METHODS
Molecular Biology
The human variant of the pore-forming α-subunit, hNaV1.5, in SP64T was graciously donated by Dr. Chris Ahern (University of British Columbia). hNaV1.5 and H880Q DNA was linearized using XbaI (Invitrogen). Transcription was completed using a Sp6 mMESSAGE mMACHINE high yield capped RNA transcription kit (Applied Biosystems, Carlsbad, CA). The C373F mutant was graciously donated by Dr. Mohamed Chahine (Laval University, Canada). C373F DNA was linearized using NotI (New England BioLabs, Pickering, Canada). C373F transcription was completed using a T7 mMESSAGE mMACHINE high yield capped RNA transcription kit (Applied Biosystems). The H880Q mutant construct was generated using the QuikChange method (Stratagene, Mississauga, Canada) with primers synthesized by Sigma Genosys (Oakville, Canada). All constructs were sequenced with the use of Eurofins MWG Operon (Huntsville, AL).
Oocyte Preparation
Oocyte preparation was completed as described previously (34). Briefly, female Xenopus laevis were terminally anesthetized using tricaine solution (2 g/liter). Oocytes were surgically removed, and theca and follicular layers were enzymatically removed by ∼1-h agitation of semi-intact lobes in a calcium-free solution, containing 96 mm NaCl, 2 mm KCl, 20 mm MgCl2, 5 mm HEPES and supplemented with 1 mg/ml type 1a collagenase. Collagenased oocytes were then washed and sorted in calcium-free solution. Stage V-VI oocytes were injected with 50 nl of cRNA encoding either WT or mutant hNaV1.5. Injected oocytes were incubated at 19 °C in SOS+ medium containing 96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 5 mm HEPES, 2.5 mm sodium pyruvate, supplemented with 100 mg/liter gentamicin sulfate and 5% horse serum for 3–10 days prior to recording. All surgical and animal care procedures were completed in accordance with the policies and procedures of the Simon Fraser University Animal Care Committee and the Canadian Council of Animal Care (35).
Data Acquisition
Data were acquired as described previously (34). Briefly, recordings were made using a CA-1B amplifier in the cut-open mode. Data were low pass-filtered at 10 kHz, digitized at 50 kHz, and then recorded using Patchmaster (HEKA Electronics, Mahone Bay, Canada) running on an iMac. Cells were permeabilized via bottom bath perfusion with intracellular solution containing 9.6 mm NaCl, 88 mm KCl, 5 mm HEPES, 11 mm EGTA, titrated to pH 7.4, and supplemented with 0.1% saponin. After 1–2-min exposure, saponin-free intracellular solution was washed in. Extracellular solution of the top and middle chambers contained 96 mm NaCl, 4 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, and 5 mm HEPES. For recordings completed below extracellular pH 6.5, HEPES was substituted with 5 mm MES. Bath chambers were temperature-controlled at 22 °C using a Peltier device run by a TC-10 temperature controller (Dagan, Minneapolis, MN).
Pulse Protocols
Cells were maintained at a holding potential of −100 mV between all protocols. Unless otherwise stated, leak subtraction was completed online, using a −p/4 protocol from a holding potential of −100 mV. Current/voltage relationships, steady-state FI (SSFI), and FI recovery and onset were measured as described previously (34).
Prolonged versions of the FI protocols were used to measure SI. Steady-state SI (SSSI) was measured by holding the membrane potential at −150 mV for 30 s to recover channels from the slow inactivated state and then stepping to 60-s conditioning pulses between −150 and −10 mV in 20-mV increments. The membrane potential was then hyperpolarized to −150 mV for 20 ms to allow recovery of fast inactivated but not slow inactivated channels and then depolarized to a 0-mV test pulse to measure the remaining channel availability. Several cells were administered a −130 mV holding potential rather than −150 mV. Changing the holding potential from −150 to −130 mV had no effect on parameters of SSSI in any of the constructs. The rate of recovery from SI was measured by depolarizing the membrane to 0 mV for 60 s to fully slow inactivate channels. The membrane potential was then stepped to an interpulse potential (−150 mV), 0.2–30 s in duration, before measuring the recovered current with a 0-mV test pulse. To measure the rate of SI onset, cells were held at −150 mV for 30 s, stepped to an prepulse potential (0 mV) for 0–60 s, hyperpolarized to −150 mV for 20 ms to allow recovery of fast inactivated but not slow inactivated channels, and then available current was measured with a 0-mV test pulse.
Use-dependent inactivation was recorded as described previously (34). Briefly, the membrane was repetitively depolarized to 0 mV for 230 ms and hyperpolarized to −90 mV for 150 ms 500 times at a frequency of ∼2.6 Hz. The frequency, which coincides with a 156-beat/min heart rate, was sufficient to generate a measurable reduction in current due to inactivation while remaining within the range of physiological depolarization and frequency values. The ratio of depolarization to repolarization was chosen based on Bazett's formula to generate a healthy QT duration, calculated QTc = 371 (36). No leak subtraction was used in use-dependent inactivation (UDI) recordings to ensure appropriate pulse frequency.
Data Analysis
Analysis of activation, FI, and SI data were completed using Fitmaster version 2x32 (HEKA Electronics) and Igor Pro version 5.01 (Wavemetrics, Lake Oswego, OR) run on an iMac. Conductance curves were computed from current/voltage relationships using the equation,
where G represents conductance, Imax represents the peak test pulse current, Vm is the test pulse voltage, and Erev is the measured reversal potential. Values were plotted as a function of test potential and then fitted with the Boltzmann equation,
where z represents the apparent valence, e0 is the elementary charge, V½ is the midpoint, T is the recording temperature in kelvin, and k is the Boltzmann constant. SSFI and SSSI data were fitted with a modified Boltzmann equation,
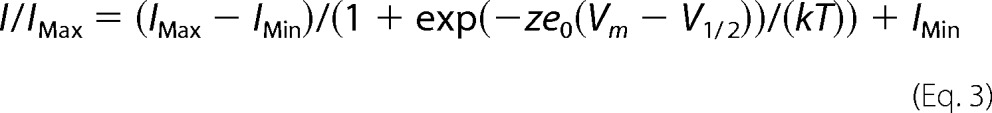 |
where IMax and IMin represent the maximum and minimum values of the fit, respectively. All other values are identical to those in Equation 2. The time constants of the recovery and onset of FI were calculated from the single exponential equation,
where Y0 represents the asymptote of the fit, A is the relative component of the exponent, τ is the time constant, and x is time. The time constants of FI onset and recovery were plotted as a function of prepulse and interpulse voltage, respectively, and fitted with the Eyring model,
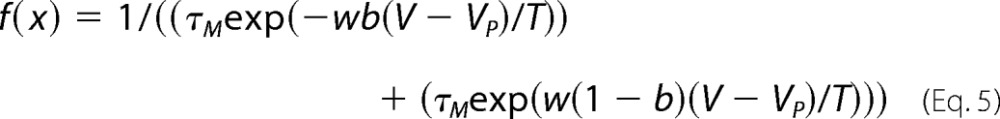 |
where τM represents the inverse of the maximum time constant, w is the reaction velocity, b is the barrier distance, V is the voltage, VP is the voltage at the peak of the fit, and T is the temperature. SI onset and recovery as well as UDI data were fitted with the double exponential equation,
where Y0 represents the asymptote of the fit, A1 is the relative component of the first exponent, τ1 is the slow time constant, A2 is the relative component of the second exponent, τ2 is the fast time constant, and x is time. Proton block data were plotted as a function of pH and were fitted with the Hill equation,
where YM and Y0 represent the maximum and minimum values of the fit, respectively, X½ is the midpoint of the curve, X is the pH, and b is the rate. Statistical analysis was completed using Student's t test and an analysis of variance, where appropriate, using JMP (SAS Institute Inc., Cary, NC). All data are reported as mean ± S.E., and statistical significance was taken at p < 0.05.
RESULTS
We sought to identify amino acid residues responsible for pH-dependent modulation of NaV1.5 channel gating. Previous studies have isolated proton sensor residues within the p-loop of several NaV and KV channels (32, 33, 37, 38). We therefore focused our efforts on the NaV1.5 channel p-loop regions.
Activation and Proton Block
Reducing extracellular pH from pH 7.4 to pH 6.0 reduces single channel conductance and significantly depolarizes the midpoint (V½) and reduces the apparent valence (z) of activation of NaV1.5 channels (32–34). In this study, the H880Q and C373F mutants displayed a significant reduction in proton block at pH 6.0 relative to WT channels (Fig. 1). At pH 6.0, the maximum channel conductance was reduced by 38.7 ± 2.0, 27.0 ± 2.6, and 19.4 ± 2.2% relative to pH 7.4 for WT, H880Q, and C373F channels, respectively (Fig. 1, B, C, and D). To assess the profile of proton block for the three constructs, maximum conductance is plotted as a function of pH and fitted with a Hill curve (Equation 7) (Fig. 1E). The pKa values were not significantly different between the three constructs: WT, pKa = 5.8 ± 0.7; H880Q, pKa = 5.7 ± 0.04; C373F, pKa = 5.9 ± 0.1. The asymptotes of proton block, however, differed significantly from zero for H880Q and C373F (6.2 ± 1.6 and 8.5 ± 1.7%, respectively, p < 0.05, n = 4) but not WT channels (0.2 ± 2.9%).
FIGURE 1.

Protons differentially block mutant NaV1.5 channels. A, sample ionic traces from WT, H880Q, and C373F channels recorded with extracellular solution titrated to pH 7.4. B–D, current/voltage (I/V) relationships from WT (B), H880Q (C), and C373F (D) channels recorded with extracellular solution titrated to pH 7.4 (solid lines) and pH 6.0 (dotted lines). Currents were normalized to the peak current amplitude recorded at pH 7.4 and plotted as a function of test potential. Proton block at pH 6.0 was significantly decreased in H880Q and C373F channels relative to WT: 27.0 ± 2.6, 19.4 ± 2.2, and 38.8 ± 1.9%, respectively (p < 0.01, n = 9–13). E, conductance, normalized to the maximum conductance for each experiment (typically pH 8.0), is plotted as a function of pH and fitted with a Hill curve (Equation 7) for WT (squares), H880Q (circles), and C373F (triangles) channels. Asymptotes based on fit lines of the mean data are displayed for H880Q (dashed line, 6.3%), and C373F (dotted line, 15.2%) channels. Based on individual fits, the asymptotes of H880Q and C373F, but not WT, were significantly elevated from zero, 6.2 ± 1.6, 8.5 ± 1.7, and 0.2 ± 2.9%, respectively (p < 0.05, n = 4). Error bars, S.E.
We have previously shown that protons shift the voltage dependence of WT NaV1.5 channel activation and SSFI and reduce the apparent valence of activation. Fig. 2 and Table 1 show that C373F and H880Q did not abolish this effect of protons. Surprisingly, the V½ of activation and SSFI in the H880Q mutant was significantly depolarized compared with C373F and WT channels, and the apparent valence of activation in both mutants was significantly greater than that in WT channels (Fig. 2). Window currents were estimated from the overlay of paired activation and SSFI curves. Peak window current occurred over a more depolarized voltage range in H880Q than C373F and WT channels: peak window current occurred at −51.3 ± 1.8, −56.9 ± 1.1, and −60.3 ± 1.0 mV for H880Q, C373F, and WT, respectively (Fig. 2, C, D, and E). We also measured proton modulation of FI recovery (−150 through −70 mV), closed state FI (−90 through −40 mV), and open state FI (−40 through +30 mV) in all three channel types (data not shown). In each case, the results reflected the previously described trends, where acidic pH causes a depolarizing shift in gating, comparable with previous findings (34, 39), and H880Q gating is depolarized relative to WT and C373F channels. These data suggest that proton modulation of channel activation and slow inactivation are two independent processes.
FIGURE 2.

Proton modulation of activation, SSFI, and window current. A, normalized conductance is plotted as a function of test potential and fitted with a Boltzmann function (Equation 2) for WT (squares), H880Q (circles), and C373F (triangles) channels. Reducing extracellular pH from pH 7.4 to pH 6.0 significantly depolarized the V½ and reduced the z of activation in WT and C373F channels but not H880Q, although all three constructs demonstrated a similar response to pH 6.0 (p < 0.05; see Table 1 for values). The V½ of the H880Q mutant was significantly depolarized compared with C373F and WT channels, −27.8 ± 1.4, −34.3 ± 1.3, and −33.6 ± 1.1 mV, respectively (p < 0.05). The z values of activation of both H880Q and C373F were significantly larger than for WT channels (4.1 ± 0.2 e, 3.9 ± 0.2 e, and 3.1 ± 0.1 e, respectively). B, proton modulation of SSFI. Normalized current is plotted as a function of prepulse potential and fitted with a Boltzmann function (Equation 3) for WT (squares), H880Q (circles), and C373F (triangles) channels. Reducing extracellular pH from pH 7.4 to pH 6.0 significantly depolarized the V½ of all three constructs but did not affect the z of SSFI. Again, the V½ of the H880Q mutant was significantly depolarized compared with C373F and WT channels at pH 7.4 but not pH 6.0 (−71.2 ± 1.2 mV, −76.3 ± 0.6 mV, and −77.3 ± 0.4 mV, respectively). C–E, activation/SSFI overlays of Boltzmann fits displaying the similar trend of proton modulation of WT (C), H880Q (D), and C373F (E) channels recorded at pH 7.4 (solid lines) and pH 6.0 (dotted lines). The window current peaks, measured from the overlay paired activation and SSFI curves, reflected the voltage dependence of activation and SSFI. Window current peaks recorded at pH 7.4 are indicated by vertical dotted lines. H880Q was significantly right-shifted relative to C373F and WT channels (−51.3 ± 1.8, −56.9 ± 1.1, and −60.3 ± 1.0 mV, respectively). The insets of A and B display the protocols used, respectively. Error bars, S.E.
TABLE 1.
Activation and fast inactivation
| Activation |
Fast inactivation |
|||||
|---|---|---|---|---|---|---|
| V½ | z | n | V½ | z | n | |
| mV | e | mV | e | |||
| pH 7.4 | ||||||
| NaV1.5 | −33.6 ± 1.1 | 3.1 ± 0.1 | 15 | −77.3 ± 0.4 | −4.6 ± 0.2 | 15 |
| H880Q | −27.8 ± 1.4a,b | 4.1 ± 0.2a | 8 | −71.2 ± 1.2a,b | −4.2 ± 0.2 | 8 |
| C373F | −34.3 ± 1.3 | 3.9 ± 0.2a | 7 | −76.3 ± 0.6 | −4.6 ± 0.1 | 9 |
| pH 6.0 | ||||||
| NaV1.5 | −27.8 ± 1.4c | 2.6 ± 0.1c | 9 | −74.5 ± 0.7c | −4.4 ± 0.3 | 10 |
| H880Q | −25.4 ± 2.1 | 3.8 ± 0.3a | 7 | −67.9 ± 2.0c | −4.0 ± 0.2 | 7 |
| C373F | −30.8 ± 3.4c | 3.2 ± 0.1a,c | 7 | −71.6 ± 0.7c | −4.6 ± 0.1 | 9 |
a p < 0.05 versus NaV1.5.
b p < 0.05 versus C373F.
c p < 0.05 versus pH 7.4.
Steady-state Slow Inactivation
Protons bind within the extracellular NaV channel pore (32, 33). Given the mutual association of protons and SI at the outer turret, we hypothesized that residues within the pore mediate proton modulation of SI. We therefore measured the steady-state and kinetic parameters of SI in all three constructs with extracellular solution titrated to either pH 7.4 or pH 6.0. Fig. 3 displays SSSI recorded in WT, C373F, and H880Q channels at pH 7.4. Normalized current was plotted as a function of prepulse potential and fitted with a Boltzmann function (Equation 3). Both H880Q and C373F channels displayed an altered response to reduced extracellular pH compared with WT channels. Extracellular perfusion of pH 6.0 triggered a significant (albeit small) increase in the maximum probability of SSSI in H880Q and C373F channels (Fig. 3, C and D, and Table 2). pH 6.0 also reduced the apparent valence of SSSI in C373F but had no effect on the apparent valence of H880Q SSSI (Fig. 3, C and D, and Table 2). In contrast, reducing extracellular pH from pH 7.4 to pH 6.0 had no effect on the SSSI curve for WT channels (Fig. 3B). This correlates with previous recordings from NaV1.5 channels expressed in oocytes (34). H880Q and C373F channels also displayed a reduced maximum probability of SSSI relative to WT channels, measured at pH 7.4 (Table 2).
FIGURE 3.

Protons differentially modulate SSSI recorded in WT, C373F, and H880Q channels. A, normalized current is plotted as a function of prepulse potential and fitted with a Boltzmann function (Equation 3) in WT (squares), C373F (triangles), and H880Q (circles). H880Q and C373F channels displayed a reduced maximum probability of SSSI relative to WT channels (see Table 2 for values). B, reducing extracellular pH from pH 7.4 (solid lines) to pH 6.0 (dotted lines) had no effect on WT SSSI. pH 6.0 significantly increased SSSI maximum probability in H880Q (C) and C373F (D) channels from 46.0 ± 1.8 to 49.5 ± 1.6% and from 45.1 ± 3.3 to 48.7 ± 3.0%, respectively (n = 5–11, p < 0.05). pH 6.0 also reduced the z of SSSI in C373F channels from −3.7 ± 0.2 e to −2.5 ± 0.2 e (n = 6, p < 0.05). The inset in A depicts the protocol used. Error bars, S.E.
TABLE 2.
Steady-state slow inactivation
| V½ | z | Maximum | n | |
|---|---|---|---|---|
| mV | e | % | ||
| pH 7.4 | ||||
| NaV1.5 | −78.0 ± 1.7 | −3.2 ± 0.3 | 55.0 ± 2.3 | 13 |
| H880Q | −79.9 ± 1.5 | −3.9 ± 0.4 | 46.0 ± 1.8a | 11 |
| C373F | −78.5 ± 1.2 | −3.7 ± 0.2 | 45.1 ± 3.3a | 6 |
| pH 6.0 | ||||
| NaV1.5 | −82.3 ± 2.8 | −2.8 ± 0.5 | 59.1 ± 4.9 | 6 |
| H880Q | −76.8 ± 3.4 | −2.8 ± 0.5 | 49.5 ± 1.6a,b | 5 |
| C373F | −75.1 ± 2.8 | −2.5 ± 0.2b | 48.7 ± 3.0a,b | 6 |
a p < 0.05 versus NaV1.5.
b p < 0.05 versus pH 7.4.
Slow Inactivation Recovery
We measured proton modulation of SI recovery at −90 mV because it correlated with the hyperpolarizing pulse of our UDI protocol (discussed below) and could therefore help explain changes in UDI between the three channel constructs. In each channel, SI recovery kinetics were biexponential. Protons accelerated SI recovery kinetics in WT but not H880Q and C373F channels (Fig. 4). In WT channels, pH 6.0 significantly increased τslow and reduced the relative contribution of the slow component. Protons did not significantly affect τfast but significantly increased the amplitude of the τfast component (Fig. 4, A (inset) and B, and Table 3). Overall, the effects on the relative amplitudes of fast and slow decay were dominant to the changes in the fast and slow time constants, and extracellular acidosis accelerated SI recovery in WT channels (Fig. 4B). In H880Q channels, protons significantly increased τfast as well as its relative component but did not affect τslow (Fig. 4, A (inset) and C, and Table 3). The C373F mutation abolished proton modulation of SI recovery (Fig. 4, A (inset) and D, and Table 3). These data demonstrate that His-880 and Cys-373 both mediate modulation of SI recovery by protons and show that Cys-373 plays a greater role in mediating the response of SI to protons than His-880. Last, the apparent differences in recovery kinetics between constructs at pH 7.4 were not statistically significant (Fig. 4A and Table 3).
FIGURE 4.

Protons differentially modulate SI recovery. A, SI recovery at −90 mV in WT (squares), H880Q (circles), and C373F (triangles) recorded at pH 7.4. Normalized current is plotted as a function of prepulse duration and fitted with a double exponential function (Equation 6). The inset of A depicts the pulse protocol used as well as bar graphs showing τfast and τslow and relative components of τfast and τslow for SI recovery. B–D, exponential fits of WT (B), H880Q (C), and C373F (D) channels at pH 7.4 (solid lines) and pH 6.0 (dotted lines). Overall, pH 6.0 accelerated SI recovery in WT (B) but had little effect on H880Q (C) and C373F (D) channels. *, statistically significant at p < 0.05. Error bars, S.E.
TABLE 3.
Slow inactivation recovery at −90 mV
| τFast | AFast | τSlow | ASlow | n | |
|---|---|---|---|---|---|
| s | % | s | % | ||
| pH 7.4 | |||||
| NaV1.5 | 1.5 ± 0.4 | 13.9 ± 2.3 | 11.4 ± 1.1 | 27.8 ± 4.2 | 7 |
| H880Q | 1.9 ± 0.2 | 20.6 ± 2.7 | 7.3 ± 0.8 | 20.0 ± 2.8 | 4 |
| C373F | 2.4 ± 0.4 | 19.6 ± 3.0 | 11.0 ± 4.0 | 17.4 ± 2.1 | 7 |
| pH 6.0 | |||||
| NaV1.5 | 2.1 ± 0.3 | 25.0 ± 4.2a,b | 25.4 ± 5.3b | 14.5 ± 2.8b | 7 |
| H880Q | 4.5 ± 0.3b | 30.8 ± 3.8b,c | 42.5 ± 27 | 19.2 ± 2.0 | 4 |
| C373F | 2.8 ± 0.5 | 19.8 ± 3.4c | 14.8 ± 3.7 | 14.9 ± 1.1 | 8 |
a p < 0.05 versus C373F.
b p < 0.05 versus pH 7.4.
c p < 0.05 versus NaV1.5.
Slow Inactivation Onset
We assessed proton modulation of SI onset at 0 mV to coincide with the depolarizing pulse of our UDI protocol and could therefore help explain changes in UDI between the three channel constructs. Protons displayed differential modulation of SI onset in WT, H880Q, and C373F channels (Fig. 5 and Table 4). SI onset was slowed in WT channels at reduced extracellular pH (Fig. 5B). WT channels displayed significantly increased τfast and τslow at pH 6.0 relative to pH 7.4 (Fig. 5, B and E, and Table 4). Extracellular perfusion of pH 6.0 did not affect the relative components of SI onset (Fig. 5F and Table 4). SI onset was also slowed at pH 6.0 in the H880Q mutant, although the effect on τfast was significantly reduced relative to WT (Fig. 5E). pH 6.0 increased τfast in WT channels by 8.0 ± 2.4 ms but increased τfast by only 1.1 ± 0.4 ms in H880Q channels. Conversely, C373F abolished proton modulation of τfast and τslow; however, pH 6.0 significantly increased the relative component of τslow (Fig. 5, D–F, and Table 4). Protons did not significantly affect the asymptote of SI onset in any of the constructs. These data demonstrate that His-880 and Cys-373 both mediate proton modulation of SI onset, and, as is the case for SI recovery, Cys-373 plays a greater role in mediating the response of SI to protons than His-880.
FIGURE 5.

Protons differentially modulate SI onset. A, normalized current amplitude is plotted as a function of prepulse duration and fitted with a double exponential equation (Equation 6) for WT (squares), H880Q (circles), and C373F (triangles) channels at pH 7.4. The asymptote of SI onset at pH 7.4 was significantly different between WT, H880Q, and C373F channels: 43.5 ± 2.2, 48.6 ± 1.2, and 57.9 ± 1.5%, respectively (Table 4). B, pH 6.0 significantly increased τast and τlow but did not affect the relative component of either time constant in WT channels. C, proton modulation of SI recovery was preserved in the H880Q mutant, although the effect on τfast was significantly reduced relative to WT (p < 0.05). D, C373F abolished proton modulation of τfast and τslow; however, pH 6.0 significantly increased the relative component of τslow. The inset in D displays the pulse protocol used. Protons did not significantly affect the asymptote of SI onset in any of the constructs. E and F, bar graphs showing τfast and τslow (E) and relative components of τfast and τslow (F) for SI recovery at pH 7.4 (filled bars) and pH 6.0 (open bars). *, statistically significant at p < 0.05. Error bars, S.E.
TABLE 4.
Slow inactivation onset at 0 mV
| τFast | AFast | τSlow | ASlow | Asymptote | n | |
|---|---|---|---|---|---|---|
| s | % | s | % | % | ||
| pH 7.4 | ||||||
| NaV1.5 | 3.0 ± 1.1 | 31.1 ± 5.0 | 35.7 ± 10.6 | 30.0 ± 5.5a | 38.5 ± 3.3a | 6 |
| H880Q | 3.2 ± 0.3 | 31.8 ± 1.6 | 27.5 ± 7.7 | 19.8 ± 0.9b | 48.6 ± 1.2a,b | 5 |
| C373F | 3.2 ± 0.3 | 26.5 ± 1.6 | 20.7 ± 4.3 | 14.7 ± 0.5b | 57.9 ± 1.5b | 6 |
| pH 6.0 | ||||||
| NaV1.5 | 10.9 ± 2.5a,c | 29.9 ± 0.8 | 76.4 ± 20.4a,c | 27.4 ± 3.8 | 41.1 ± 2.7a | 6 |
| H880Q | 4.3 ± 0.5c | 33.2 ± 1.2 | 42.6 ± 8.5b,c | 18.8 ± 2.7 | 49.1 ± 2.6 | 5 |
| C373F | 3.0 ± 0.9b | 21.6 ± 3.5 | 19.4 ± 3.7b | 22.8 ± 2.5c | 55.5 ± 1.5b | 7 |
a p < 0.05 versus C373F.
b p < 0.05 versus NaV1.5.
c p < 0.05 versus pH 7.4.
The asymptote of SI onset at pH 7.4 was significantly different between the WT, H880Q, and C373F constructs (Fig. 5A and Table 4). Additionally, H880Q and C373F channels displayed a reduced τslow component relative to WT channels (Table 4). The differences in the SI onset asymptote between channel constructs are probably attributable to the observed reductions in the components of τslow in H880Q and C373F.
Use-dependent Inactivation
Protons display profound isoform-specific modulation of UDI (39). UDI in NaV1.4 is pH-insensitive, whereas in NaV1.5, UDI is destabilized by protons. Inserting the p-loops of the skeletal muscle isoform, NaV1.4, into the NaV1.5 channel backbone abolishes proton modulation of UDI (40). We measured UDI using a pulse protocol designed to mimic a human ventricular action potential, as described previously (34). Cells were depolarized to 0 mV for 230 ms and then hyperpolarized to −90 mV for 150 ms to simulate an elevated heart rate of ∼160 beats/min. This stimulation frequency is high enough to induce readily measurable levels of UDI but low enough to remain physiologically relevant. Peak currents from 500 consecutive pulses were recorded and normalized to the first pulse and then plotted as a function of time and fitted with a double exponential equation (Equation 6) as in Fig. 6.
FIGURE 6.

Turret mutations abolish proton modulation of UDI. A–C, normalized current is plotted as a function of time and fitted with a double exponential function (Equation 6). Error bars are omitted for image clarity. A, pH 6.0 (gray symbols) significantly increased the τFast from 4.0 ± 0.5 to 7.2 ± 0.7 s and the asymptote of UDI from 60.6 ± 1.1 to 65.3 ± 1.4% in WT channels. B, the C373F mutation removed proton modulation of the UDI asymptote but not τFast. pH 6.0 significantly increased τFast in C373F channels from 2.5 ± 0.2 to 5.0 ± 0.6 s. Additionally, τFast in C373F channels was significantly reduced compared with WT channels (Table 5). C, UDI in H880Q channels was not modulated by protons. D and E, bar graphs depicting τFast (D) and asymptote (E) of UDI of WT, C373F, and H880Q channels at pH 7.4 (filled bars) and pH 6.0 (open bars). The inset of B displays the pulse protocol used. *, statistically significant at p < 0.05. Error bars, S.E.
In WT channels (Fig. 6A), we observed the previously described destabilization of UDI in response to reduced extracellular pH (34, 39). WT channels displayed a significant increase in the asymptote of UDI at pH 6.0 relative to pH 7.4 (Fig. 6A and Table 5). The C373F mutation removed the effects of protons on the asymptote of UDI but preserved proton modulation of τFast (Fig. 6C). C373F channels also displayed a significantly reduced τFast compared with WT (Table 5). H880Q completely abolished the effects of protons on UDI (Fig. 6, B, D, and E, and Table 5).
TABLE 5.
Use-dependent inactivation
| τFast | AFast | τSlow | ASlow | Asymptote | n | |
|---|---|---|---|---|---|---|
| s | % | s | % | % | ||
| pH 7.4 | ||||||
| NaV1.5 | 4.0 ± 0.5 | 23.7 ± 2.5a | 38.5 ± 7.1 | 14.7 ± 1.0 | 60.6 ± 1.1a | 9 |
| H880Q | 3.5 ± 0.4 | 24.9 ± 2.2a | 36.7 ± 10.8 | 9.4 ± 1.3b | 64.1 ± 1.8a | 6 |
| C373F | 2.5 ± 0.2b | 16.1 ± 0.8b | 36.0 ± 16.5 | 9.8 ± 1.3b | 72.2 ± 1.5b | 8 |
| pH 6.0 | ||||||
| NaV1.5 | 7.2 ± 0.7c | 22.1 ± 0.4 | 61.4 ± 11.5 | 15.3 ± 0.7 | 65.3 ± 1.4c | 8 |
| H880Q | 4.6 ± 0.7 | 28.1 ± 3.4a | 58.7 ± 7.7 | 11.4 ± 1.4 | 59.3 ± 4.0 | 6 |
| C373F | 5.0 ± 0.6c | 19.7 ± 2.0 | 57.5 ± 16.6 | 9.4 ± 1.9b | 69.8 ± 2.4 | 6 |
a p < 0.05 versus C373F.
b p < 0.05 versus NaV1.5.
c p < 0.05 versus pH 7.4.
Last, both C373F and H880Q significantly reduced the relative components of τSlow compared with WT under control conditions (Table 5). The C373F mutant also displayed a significantly elevated UDI asymptote compared with WT and H880Q channels (Fig. 6 and Table 3). These data correlate well with the reduced maximum probability of SI observed in C373F and H880Q channels (Figs. 4 and 5).
DISCUSSION
Proton block and modulation of several NaV1.5 gating parameters occurs with a pKa similar to a histidine (pKa ∼6.0) (32, 34). p-loop histidines are proton sensors in voltage-gated potassium channels, whereby replacement with a glutamine abolishes proton modulation of C-type inactivation (38, 41). There are two p-loop histidines in NaV1.5, His-880 and His-886. We created the NaV1.5 mutants H880Q and H886Q, simulating constitutive deprotonation at those positions. The H886Q mutant, however, did not produce measurable currents. Cys-373 was shown previously to be involved in NaV1.5 proton block and seemed a likely candidate to modulate slow inactivation (32). We thus recorded ionic currents from three NaV1.5 channel constructs: WT and two p-loop mutants, C373F and H880Q. We found that H880Q shifted the voltage dependence of activation relative to WT channels, reduced the pH-dependent modulation of slow inactivation kinetics, and abolished the pH dependence of UDI. C373F did not affect activation but removed the pH-dependent modulation of SI kinetics and reduced the pH dependence of UDI. Last, both mutations reduced proton block and destabilized SI relative to WT channels.
Molecular Determinants of Proton Block
Unlike NaV1.5, the skeletal muscle isoform, NaV1.4, is not fully blocked by protons (32, 33). NaV1.4 channels display a pH-insensitive current that is roughly ∼14% of maximum conductance (33). In NaV1.4, replacement of Tyr-402 with a cysteine (the homologous residue in NaV1.5) abolishes the proton-insensitive current (32). The reverse mutation in NaV1.5, C373Y, imparts a proton-insensitive current similar to that seen in NaV1.4 (32). Replacement of Tyr-402 with a phenylalanine or a serine, the equivalent residues in NaV1.2 and NaV1.8, respectively, does not alter the pH-insensitive current, suggesting that NaV1.2 and NaV1.8 would have a proton block asymptote similar to that of NaV1.4 (32, 39). Kahn et al. (32) suggested that protonation of Cys-373 along with the outer pore carboxylates Glu-375, Glu-901, Asp-1423, and Asp-1714 imparts proton block in NaV1.5 and that the absence of this pore cysteine in other NaV channels results in a pH-insensitive current.
Here we report that two novel mutations, C373F and H880Q, alter NaV1.5 channel proton block. Both mutations significantly reduced the degree of proton block observed at pH 6.0 relative to WT channels and displayed asymptotes of proton block that represented a pH-insensitive component of current (Fig. 1). C373F proton block data fitted with a Hill curve (Equation 7) displayed a pH-insensitive conductance that was roughly 9% of maximum conductance (Fig. 1E), consistent with previous results from the C373Y NaV1.5 mutant (32). H880Q also displayed a proton block asymptote that was significantly different from zero at ∼6% of maximum conductance (Fig. 1E). His-880 has not been implicated previously in NaV channel proton block, and it is interesting that H880Q proton block was intermediate between that observed in C373F and WT channels. Kahn et al. (33) hypothesized that protonation at the p-loop increases the electrostatic potential within the NaV channel outer vestibule, thereby repelling Na+ ions. Our results further support that hypothesis. Cys-373 is adjacent to the selectivity filter, where the electrostatic potential is the most negative. His-880, however, is predicted to be at the top of the NaV channel P1 helix of DII (Fig. 7). Based on their relative locations, it is understandable that the effect of the H880Q mutation would be less dramatic than that observed by the C373F mutation.
FIGURE 7.

A, predicted NaV channel secondary structure depicting the relative locations of the C373F (★) and H880Q (♦) mutations. B, sequence alignment of the p-loop segments in bacterial NaChBac, NaVAb, and DI and DII of the mammalian NaV1.2 (accession number Q99250.3) and NaV1.5 (accession number Q14524.2) channels (1). Selectivity residues of NaChBac and NaVAb along with the homologous residues in NaV1.2 and NaV1.5 channels are highlighted in gray. Residues that differ between NaV1.2 and NaV1.5 are encapsulated. P1 and P2 represent the two pore helices isolated in the crystal structure of the NaVAb channel (17, 43).
Pore Mutants Destabilize Slow Inactivation
NaV1.2 channels inactivate more completely than NaV1.5; during prolonged depolarization, more than 65% of NaV1.2 channels slow inactivate compared with ∼50% in NaV1.5 (8, 14, 34, 42). Because the homologous position to NaV1.5 Cys-373 is Phe-385 in NaV1.2 channels, and because SI is known to be sensitive to mutations within the external pore, we predicted that the C373F mutant would confer a stabilization of slow inactivated states in Nav1.5 (Fig. 7) (8, 14, 32). Because His-880 is highly conserved (Fig. 7), we also predicted that the H880Q mutation would disrupt NaV1.5 SI. Interestingly, both H880Q and C373F destabilized the slow inactivated state. The maximum probability of SSSI was reduced by ∼10% in H880Q and C373F channels relative to WT (Table 2). Additionally, compared with WT channels, the asymptote of SI onset measured at 0 mV was increased in H880Q and C373F channels by ∼10 and ∼19%, respectively (Table 4). Destabilization of SI by C373F was further implicated by an observed increase in the asymptote of UDI, ∼12% (Table 5). The asymptote of H880Q UDI displayed an elevated trend (∼4%), but the difference from WT was not statistically significant (Table 5).
Although alterations to SI by H880Q were predicted because His-880 is highly conserved throughout mammalian NaV isoforms, destabilized SI in C373F channels was surprising. As mentioned previously, Cys-373 in NaV1.5 is homologous to Phe-385 in NaV1.2 (Fig. 7), and, because NaV1.2 has more complete SI than NaV1.5, C373F was expected to enhance SI (8, 14, 34, 42). In contrast to expectations, C373F reduced the maximum probability of SI. When Ile-891, positioned in the NaV1.5 p-loop (Fig. 7), is replaced with a valine, the homologous residue in NaV1.4, the mutant channel displays NaV1.4-like SI (14). The reverse mutation in NaV1.4 channels confers NaV1.5-like SI (14). NaV1.2 also has a valine at this position (Val-935; Fig. 7). Our data suggest that the differences in maximum probability of SI between NaV1.5 and NaV1.2 channels may not be isolated to a single residue as is the case between NaV1.5 and NaV1.4.
Proton Sensors and Slow Inactivation
We measured UDI using a pulse protocol designed to mimic a human ventricular action potential cycling at 2.6 Hz or ∼160 beats/min, as described previously (34). Given the length of the depolarizing and hyperpolarizing pulses (230 and 150 ms, respectively), this protocol is an effective tool to assess the dynamic equilibrium of slow inactivated states, which have onset and recovery time constants in the range of seconds to tens of seconds (Tables 3 and 4). Fast inactivation, with onset and recovery time constants less than 30 ms, has a negligible contribution to the decay of current during this experimental protocol (34). Our data demonstrate that p-loop residues mediate proton modulation of UDI of NaV1.5 channels.
Both H880Q and C373F abolished proton modulation of UDI. Protons increase, reduce, or have no effect on the asymptote of UDI in NaV1.5, NaV1.2, and NaV1.4 channels, respectively (34, 39). Inserting the p-loops of NaV1.4 into the NaV1.5 channel backbone imparts NaV1.4 channel-like proton modulation of UDI (40). The p-loops between isoforms are highly conserved, and Cys-373 is one of only a few positions that differ between the three channels. As mentioned previously, Cys-373 is responsible for isoform-specific proton block between cardiac and skeletal channels (32). The homologous residues in NaV1.2, NaV1.4, and NaV1.8 channels (phenylalanine, tyrosine, and serine, respectively) impart a proton-insensitive current in NaV1.4, whereas cysteine abolishes it (32). We therefore postulate that protonation of Cys-373 modulates UDI in NaV1.5 and contributes to the differences in UDI modulation between cardiac and skeletal channel isoforms. Further, our results with H880Q and C373F suggest that accumulation of positive charge around the NaV1.5 channel pore mediates destabilization of UDI and that Cys-373 protonation represents a “tipping point” of positive charge that induces the reduction in NaV1.5 UDI. These hypotheses require the assumption that SI disruption under control conditions in H880Q and C373F relative to WT is allosteric and not electrostatic.
These data paired well with our data on the kinetics of SI. C373F was more effective than H880Q at abolishing the pH-dependent modulation of SI kinetics (Figs. 4 and 5 and Tables 3 and 4). SSSI in C373F also displayed a greater response to protons than H880Q, whereas WT channels showed no change (Fig. 3 and Table 2). Overall, H880Q showed intermediate responses to protons compared with WT and C373F channels (Tables 2–4). Like their role in proton block, the relative positions of His-880 and Cys-373 may underlie the differential contribution to SI proton modulation. Because slow inactivation is thought to involve a structural rearrangement at or around the selectivity filter, protonation of Cys-373 might be predicted to have a greater effect on SI than His-880, consistent with our results.
Conclusion
Our results demonstrate that p-loop residues His-880 and Cys-373 mediate NaV1.5 sensitivity to protons. These results also identified Cys-373 as a residue responsible for isoform-specific proton modulation of UDI. It seems likely that proton block and SI modulation occur in tandem, given the strong overlap in results between the two processes. Additionally, these results suggest that His-880 and Cys-373 contribute to the stability of SI in NaV1.5 channels.
Acknowledgment
We thank Stanislav Sokolov for critical reading of the manuscript.
This work was supported in part by Natural Sciences and Engineering Research Council Discovery Grants 611509 (to P. C. R.) and 611527 (to T. W. C.) and Canadian Foundation for Innovation Leaders Opportunity Fund Project 17976 (to T. W. C. and P. C. R.).
- NaV
- voltage-gated sodium
- FI
- fast inactivation
- SI
- slow inactivation
- SSFI
- steady-state FI
- SSSI
- steady-state SI.
REFERENCES
- 1. Gellens M. E., George A. L., Jr., Chen L. Q., Chahine M., Horn R., Barchi R. L., Kallen R. G. (1992) Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc. Natl. Acad. Sci. U.S.A. 89, 554–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Makita N., Bennett P. B., George A. L. (1996) Molecular determinants of B1 subunit-induced gating modulation in voltage-dependent Na+ channels. J. Neurosci. 16, 7117–7127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Noda M. (1993) Structure and function of sodium channels. Ann. N.Y. Acad. Sci. 707, 20–37 [DOI] [PubMed] [Google Scholar]
- 4. Jensen M. Ø., Jensen T. R., Kjaer K., Bjørnholm T., Mouritsen O. G., Peters G. H. (2002) Orientation and conformation of a lipase at an interface studied by molecular dynamics simulations. Biophys. J. 98, 111a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Terlau H., Heinemann S. H., Stühmer W., Pusch M., Conti F., Imoto K., Numa S. (1991) Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 293, 93–96 [DOI] [PubMed] [Google Scholar]
- 6. Patton D. E., West J. W., Catterall W. A., Goldin A. L. (1992) Amino acid residues required for fast Na+-channel inactivation. Charge neutralizations and deletions in the III-IV linker. Proc. Natl. Acad. Sci. U.S.A. 89, 10905–10909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vilin Y. Y., Ruben P. C. (2001) Slow inactivation in voltage-gated sodium channels. Molecular substrates and contributions to channelopathies. Cell Biochem. Biophys. 35, 171–190 [DOI] [PubMed] [Google Scholar]
- 8. Vilin Y. Y., Makita N., George A. L., Jr., Ruben P. C. (1999) Structural determinants of slow inactivation in human cardiac and skeletal muscle sodium channels. Biophys. J. 77, 1384–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Todt H., Dudley S. C., Jr., Kyle J. W., French R. J., Fozzard H. A. (1999) Ultra-slow inactivation in u1 Na+ channels is produced by a structural rearrangement of the outer vestibule. Biophys. J. 76, 1335–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Balser J. R., Nuss H. B., Chiamvimonvat N., Pérez-Garcia M. T., Marban E., Tomaselli G. F. (1996) External pore residue mediates slow inactivation in u1 rat skeletal muscle sodium channels. J. Physiol. 494, 431–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Townsend C., Horn R. (1997) Effect of alkali metal cations on slow inactivation of cardiac Na+ channels. J. Gen. Physiol. 110, 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xiong W., Li R. A., Tian Y., Tomaselli G. F. (2003) Molecular motions of the outer ring of charge of the sodium channel. Do they couple to slow inactivation? J. Gen. Physiol. 122, 323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ong B. H., Tomaselli G. F., Balser J. R. (2000) A structural rearrangement in the sodium channel pore linked to slow inactivation and use dependence. J. Gen. Physiol. 116, 653–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vilin Y. Y., Fujimoto E., Ruben P. C. (2001) A single residue differentiates between human cardiac and skeletal muscle Na+ channel slow inactivation. Biophys. J. 80, 2221–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cuello L. G., Jogini V., Cortes D. M., Perozo E. (2010) Structural mechanism of C-type inactivation in K+ channels. Nature 466, 203–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cuello L. G., Jogini V., Cortes D. M., Pan A. C., Gagnon D. G., Dalmas O., Cordero-Morales J. F., Chakrapani S., Roux B., Perozo E. (2010) Structural basis for the coupling between activation and inactivation gates in K+ channels. Nature 466, 272–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Payandeh J., Gamal El-Din T. M., Scheuer T., Zheng N., Catterall W. A. (2012) Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486, 135–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang X., Ren W., DeCaen P., Yan C., Tao X., Tang L., Wang J., Hasegawa K., Kumasaka T., He J., Wang J., Clapham D. E., Yan N. (2012) Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486, 130–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Featherstone D. E., Richmond J. E., Ruben P. C. (1996) Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys. J. 71, 3098–3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McCollum I. J., Vilin Y. Y., Spackman E., Fujimoto E., Ruben P. C. (2003) Negatively charged residues adjacent to IFM motif in the DIII-DIV linker of hNaV1.4 differentially affect slow inactivation. FEBS Lett. 552, 163–169 [DOI] [PubMed] [Google Scholar]
- 21. Bendahhou S., Cummins T. R., Kula R. W., Fu Y. H., Ptácek L. J. (2002) Impairment of slow inactivation as a common mechanism for periodic paralysis in DIIS4-S5. Neurology 58, 1266–1272 [DOI] [PubMed] [Google Scholar]
- 22. Hayward L. J., Brown R. H., Jr., Cannon S. C. (1997) Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophys. J. 72, 1204–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang S. Y., Wang G. K. (1997) A mutation in segment I-S6 alters slow inactivation of sodium channels. Biophys. J. 72, 1633–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cummins T. R., Sigworth F. J. (1996) Impaired slow inactivation in mutant sodium channels. Biophys. J. 71, 227–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bendahhou S., Cummins T. R., Tawil R., Waxman S. G., Ptacek L. J. (1999) Activation and inactivation of the voltage-gated sodium channel. Role of segment S5 revealed by a novel hyperkalaemic periodic paralysis mutation. J. Neurosci. 19, 4762–4771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fleig A., Fitch J. M., Goldin A. L., Rayner M. D., Starkus J. G., Ruben P. C. (1994) Point mutations in IIS4 alter activation and inactivation of rat brain IIA Na channels in Xenopus oocyte macropatches. Pflugers Arch. 427, 406–413 [DOI] [PubMed] [Google Scholar]
- 27. Kuzmenkin A., Muncan V., Jurkat-Rott K., Hang C., Lerche H., Lehmann-Horn F., Mitrovic N. (2002) Enhanced inactivation and pH sensitivity of Na+ channel mutations causing hypokalaemic periodic paralysis type II. Brain 125, 835–843 [DOI] [PubMed] [Google Scholar]
- 28. Mitrovic N., George A. L., Jr., Horn R. (2000) Role of domain 4 in sodium channel slow inactivation. J. Gen. Physiol. 115, 707–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Capes D. L., Arcisio-Miranda M., Jarecki B. W., French R. J., Chanda B. (2012) Gating transitions in the selectivity filter region of a sodium channel are coupled to the domain IV voltage sensor. Proc. Natl. Acad. Sci. U.S.A. 109, 2648–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nguyen-Thi A., Ruiz-Ceretti E., Schanne O. F. (1981) Electrophysiologic effects and electrolyte changes in total myocardial ischemia. Can. J. Physiol. Pharmacol. 59, 876–883 [DOI] [PubMed] [Google Scholar]
- 31. Antzelevitch C., Belardinelli L., Zygmunt A. C., Burashnikov A., Di Diego J. M., Fish J. M., Cordeiro J. M., Thomas G. (2004) Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation 110, 904–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khan A., Kyle J. W., Hanck D. A., Lipkind G. M., Fozzard H. A. (2006) Isoform-dependent interaction of voltage-gated sodium channels with protons. J. Physiol. 576, 493–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khan A., Romantseva L., Lam A., Lipkind G., Fozzard H. A. (2002) Role of outer ring carboxylates of the rat skeletal muscle sodium channel pore in proton block. J. Physiol. 543, 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jones D. K., Peters C. H., Tolhurst S. A., Claydon T. W., Ruben P. C. (2011) Extracellular proton modulation of the cardiac voltage-gated sodium channel, NaV.15. Biophys. J. 101, 2147–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Drummond G. B. (2009) Reporting ethical matters in the Journal of Physiology. Standards and advice. J. Physiol. 587, 713–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bazett H. C. (1920) The time relations of the blood-pressure changes after excision of the adrenal glands, with some observations on blood volume changes. J. Physiol. 53, 320–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Claydon T. W., Boyett M. R., Sivaprasadarao A., Ishii K., Owen J. M., O'Beirne H. A., Leach R., Komukai K., Orchard C. H. (2000) Inhibition of the K+ channel kv1.4 by acidosis. Protonation of an extracellular histidine slows the recovery from N-type inactivation. J. Physiol. 526, 253–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Claydon T. W., Boyett M. R., Sivaprasadarao A., Orchard C. H. (2002) Two pore residues mediate acidosis-induced enhancement of C-type inactivation of the Kv1.4 K+ channel. Am. J. Physiol. Cell Physiol. 283, C1114–C1121 [DOI] [PubMed] [Google Scholar]
- 39. Vilin Y. Y., Peters C. H., Ruben P. C. (2012) Acidosis differentially modulates inactivation in Nav1.2, Nav1.4, and Nav1.5 channels. Front. Pharmacol. 3, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vilin Y., Ruben P. C. (2010) Differential pH-dependent regulation of NaV channels. Biophys. J. 111a [Google Scholar]
- 41. Kehl S. J., Eduljee C., Kwan D. C., Zhang S., Fedida D. (2002) Molecular determinants of the inhibition of human Kv1.5 potassium currents by external protons and Zn2+. J. Physiol. 541, 9–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Y., Yu F. H., Surmeier D. J., Scheuer T., Catterall W. A. (2006) Neuromodulation of Na+ channel slow inactivation via cAMP-dependent protein kinase and protein kinase C. Neuron 49, 409–420 [DOI] [PubMed] [Google Scholar]
- 43. Payandeh J., Scheuer T., Zheng N., Catterall W. A. (2011) The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]