

Summary
The infectivity and persistence of Mycobacterium tuberculosis requires the utilization of host cell cholesterol. We have examined the specific role of cytochrome P450 CYP125A1 in the cholesterol degradation pathway using genetic, biochemical, and high resolution mass spectrometric approaches. The analysis of lipid profiles from cells grown on cholesterol revealed that CYP125A1 is required to incorporate the cholesterol side chain carbon atoms into cellular lipids, as evidenced by an increase in the mass of the methyl-branched phthiocerol dimycocerosates. We observed that cholesterol-exposed cells lacking CYP125A1 accumulate cholest-4-en-3-one, suggesting that this is a physiological substrate for this enzyme. Reconstitution of enzymatic activity with spinach ferredoxin and ferredoxin reductase revealed that recombinant CYP125A1 indeed binds both cholest-4-en-3-one and cholesterol, efficiently hydroxylates both of them at C-27, and then further oxidizes 27-hydroxycholest-4-en-3-one to cholest-4-en-3-one-27-oic acid. We determined the x-ray structure of cholest-4-en-3-one-bound CYP125A1 at a resolution of 1.58 Å. CYP125A1 is essential for growth of CDC1551 in media containing cholesterol or cholest-4-en-3-one. In its absence, the latter compound is toxic for both CDC1551 and H37Rv when added with glycerol as a second carbon source. CYP125A1 is a key enzyme in cholesterol metabolism and plays a crucial role in circumventing the deleterious effect of cholest-4-en-3-one.
Keywords: P450, CYP125A1, cholesterol degradation, cholest-4-en-3-one toxicity, x-ray structure
Introduction
Mycobacterium tuberculosis is a successful pathogen that latently infects nearly one-third of the world’s population and causes about 2 million deaths annually. M. tuberculosis has again become a global threat due in part to the appearance of extremely drug- resistant strains that are virtually untreatable ((WHO), 2009). This makes the development of new therapeutics highly desirable, which requires the discovery and characterization of new mycobacterial targets.
The persistence mechanisms that allow M. tuberculosis to adapt and replicate in the hostile and nutrient-limited environment of the phagosome-like compartment of macrophages remain largely unknown. One persistence mechanism of M. tuberculosis is its ability to respond to oxygen deprivation by activating the devS/devR regulon (Voskuil et al., 2004, Sherman et al., 2001). Its ability to persist also depends on the ability of mycobacteria to use nutrients from the host. M. tuberculosis is known to switch its metabolic pathways to lipids rather than carbohydrates during infection (Boshoff and Barry, 2005a, Boshoff and Barry, 2005b, Schnappinger et al., 2003). Sequencing of the M. tuberculosis genome revealed at least 250 genes predicted to be involved in lipid metabolism (Cole et al., 1998), some of which have been shown to be required for infection (Brzostek et al., 2007, Brzostek et al., 2009, Chang et al., 2007, Chang et al., 2009, Hu et al., 2010, Munoz-Elias and McKinney, 2005, Yam et al., 2009).
Numerous M. tuberculosis genes required for survival in macrophages were identified by the screening of transposon mutants that failed to grow within primary macrophages (Rengarajan et al., 2005). Among these genes an operon, named igr for intracellular growth, was shown to be required for growth in macrophages and mice (Chang et al., 2007). The six genes of the igr operon (Fig. 1A), consisting of a cytochrome P450 (cyp125A1), two acyl-CoA dehydrogenases (fadE28 and fadE29), two conserved hypothetical proteins (Rv3541-2c), and a putative lipid carrier protein (ltp2), are predicted to play roles in lipid metabolism. The orthologues of these genes were also identified as part of a large genomic cluster required for the growth of Rhodococcus jostii RHA1 on cholesterol (Van der Geize et al., 2007). The participation of the igr operon in cholesterol metabolism in M. tuberculosis was reported (Chang et al., 2009) and a C27-steroid monooxygenase activity for CYP125A1 in vitro was very recently demonstrated (Capyk et al., 2009, McLean et al., 2009).
Fig. 1.
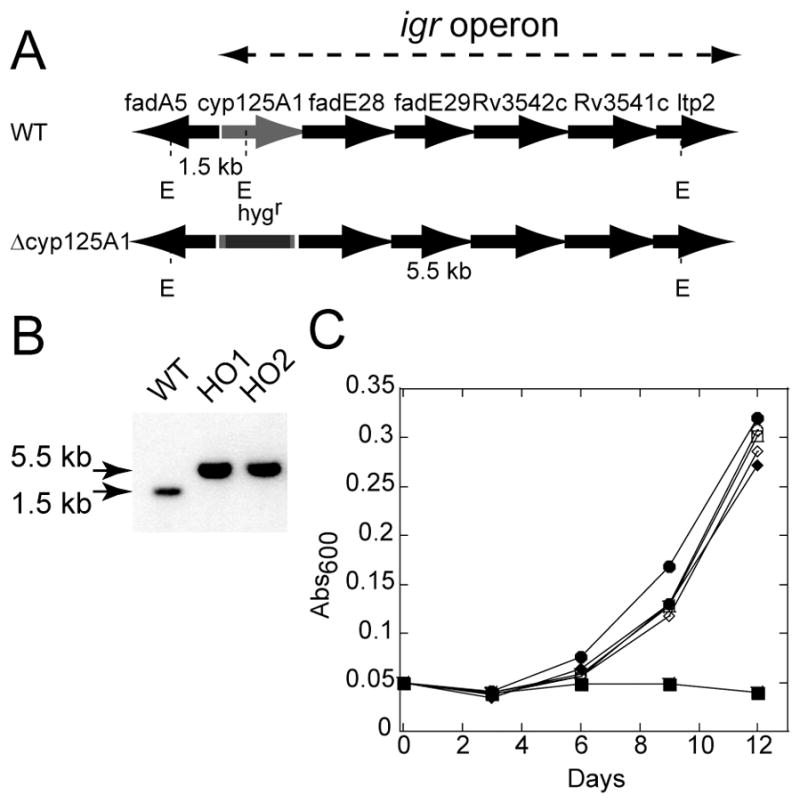
Inactivation of cyp125A1 and growth characteristics. (A) Schematic representation of the igr operon consisting of cyp125A1 and five other genes before and after insertion of the hyg resistance gene in cyp125A1. (B) Southern blot of EcoR I-digested genomic DNA showing the hybridizing fragment of WT (1.5 kb) and of two cyp125A1 deletion mutants, HO1 and HO2 (5.5 kb). (C) Representative growth curves are shown of WT (circle), Δcyp125A1 (HO1, square), and Δcyp125A1/cyp125A1 (HO11, diamond) strains on minimal media containing glycerol (open symbols) or cholesterol (filled symbols). The growth was monitored by measuring the absorbance at 600 nm as a function of time.
In this work, we addressed the role of CYP125A1 in the metabolism of cholesterol in M. tuberculosis CDC1551 cells. We used a mycobacteriophage-based transduction method to produce a deletion strain that was characterized with respect to both growth and cholesterol transformation and incorporation using high resolution mass spectrometry. The enzymatic activities were reconstituted in vitro using heterologously expressed and purified CYP125A1, and the reaction products were identified. The novelty of this work includes (1) identification of aldehyde and carboxylic acid derivatives among the products of the catalytic reaction, (2) characterization of the cellular role of CYP125A1 in alleviation of the cholest-4-en-3-one toxicity in M. tuberculosis CDC1551 cells, and (3) determination of the first x-ray structure of the CYP125A1-substrate complex using cholest-4-en-3-one as the substrate.
Results
CYP125A1 is essential for growth of M. tuberculosis CDC1551 on cholesterol
In order to determine whether CYP125A1 is required for growth on cholesterol, we produced a gene knockout in the clinically relevant M. tuberculosis CDC1551 strain. The H37Rv strain has been passaged for many decades outside of the human host. Thus, the relevance of this strain to clinical isolates has been questioned (Fleischmann et al., 2002), and there are concerns that this may have attenuated its virulence in humans (Jacobs et al., 1996). In contrast, CDC1551 is a recent clinical isolate that was responsible for an outbreak of tuberculosis in the United States in 1995 (Valway et al., 1998). Although CDC1551 was shown to induce a more vigorous host response in vivo and in vitro, it is not more virulent than other clinical isolates (Manca et al., 1999).
The disruption of cyp125A1 by deletion in CDC1551 was confirmed by Southern blot analysis. As expected for allelic exchange mutants clones were found to present a single hybridizing EcoRI fragment of 5.5 kb, whereas wild-type (WT) gave a 1.5 kb fragment (Fig. 1B). The longer hybridizing EcoRI fragment resulted from the addition of the hyg gene and the loss of an internal EcoRI site. The cyp125A1 knockout strain used in this study was named HO1.
The growth of the wild-type and knockout strains was tested in minimal media containing glycerol or cholesterol as a sole source of carbon. As shown in Fig. 1C, all strains exhibited identical growth curves with glycerol. However, HO1 cells failed to grow in the presence of cholesterol, whereas the growth of the WT strain was slightly better than with glycerol. Finally, the growth phenotype on cholesterol for the HO1 cells could almost be completely reversed when a single copy of the cyp125A1 gene was integrated into the chromosome. The complemented strain was named HO11.
Incorporation of cholesterol into methyl-branched lipids
Using a mass spectrometric approach to simultaneously monitor hundreds of lipids, it was recently discovered that the size and abundance of two methyl-branched containing lipid virulence factors, phthiocerol dimycocerosate (PDIM) and sulfolipid-1, are controlled by the availability of a common precursor, methylmalonyl CoA (Jain et al., 2007, Yang et al., 2009). M. tuberculosis was shown to use odd-chain fatty acids and to degrade cholesterol to generate a high metabolic flux of methylmalonyl CoA via the formation of propionyl-CoA. We therefore used the mass of PDIM as a measure of the incorporation of cholesterol into HO1 cells. The WT, HO1 and HO11 strains were incubated for 24 h in liquid 7H9 medium supplemented with glycerol or cholesterol. The apolar lipids were extracted and analyzed by LTQFT/MS. As shown in Fig. 2, an increase in the average mass of the PDIM was observed in the wild-type and the complemented strains, whereas no shift was observed in HO1 cells. When WT cells were incubated with heptadeuterated cholesterol, in which the hydrogen atoms of side chain positions C-25 to C-27 are replaced with deuterium, the labeled portion of the side chain was incorporated into PDIM, as evidenced by a further increase in their average mass. As expected from the study by Jain et al. (Jain et al., 2007), incubation with propionic acid also resulted in an increased mass of PDIM in both WT and HO1 (data not shown), ruling out the possibility that the lack of incorporation of the cholesterol side chain was due to a problem in PDIM biosynthesis in HO1 cells. Altogether these results demonstrate that CYP125A1 is essential for degradation of the cholesterol side chain, generating most likely a higher metabolic flux of methylmalonyl CoA.
Fig. 2.
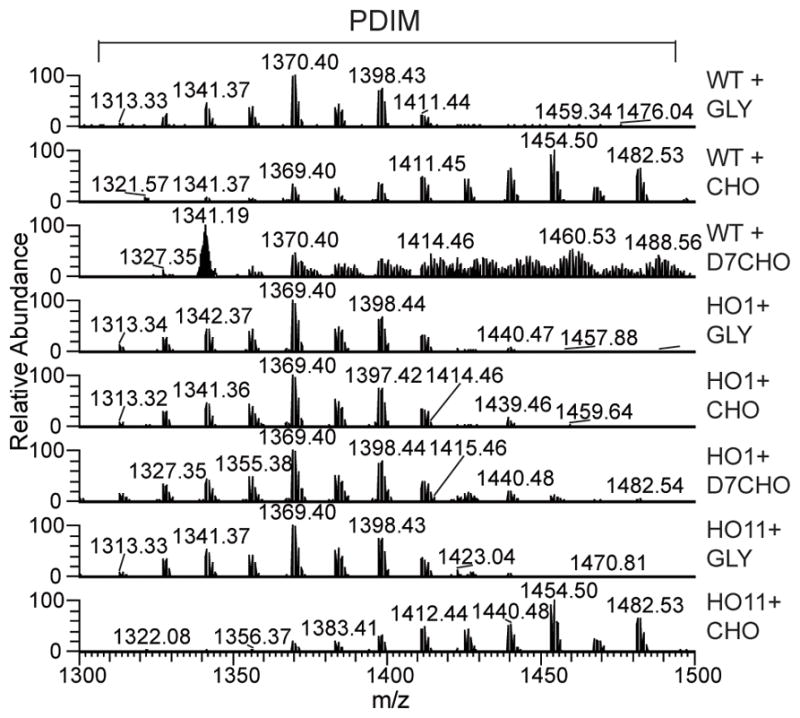
Mass spectrometric analysis of PDIM extracted from the WT, HO1 and HO11 strains. Cells were incubated with glycerol, cholesterol or heptadeuterated-cholesterol for 24 h in 7H9 medium supplemented with 0.5% bovine serum albumin, 0.085% NaCl and 0.05% Tween-80. Apolar lipids were prepared and monitored by LTQ-FT/MS as described in the Experimental Procedures section. Addition of cholesterol leads to an increase in the mass of the PDIM.
A cholesterol-derived metabolite accumulates in HO1 cells
Lipidomic experiments revealed that a metabolite with an m/z value of 385.35 accumulated to a greater extent in HO1 cells than in the WT strain. In order to identify this metabolite and to determine its relative concentration in the two strains, we separately incubated WT cells with cholesterol and HO1 cells with heptadeuterated cholesterol, and vice versa. The apolar lipids extracted from the different incubations were co-infused as 1:1 mixtures into the LTQFT/MS instrument. Approximately 40 times more cholesterol-derived metabolite with an m/z value of 392.39, corresponding to the heptadeuterated form of the 385.35-mz metabolite, accumulated in the HO1 cells (Fig. 3A (top panel)). Conversely, the metabolite (385.35-m/z) accumulated to a greater extent in cholesterol-grown HO1 cells, but the 392.39-m/z heptadeuterated metabolite was easily detected in the WT cells (Fig. 3A, middle panel), suggesting that the side chain degradation activity is subject to an isotope effect.
Fig. 3.

Accumulation of a cholesterol-derived metabolite in M. tuberculosis cells. (A) Co-injection (1:1) of apolar lipid extracts prepared from WT and HO1 cells incubated with cholesterol and heptadeuterated-cholesterol, respectively (top panel), and vice-versa (bottom panel). The m/z value for the non-deuterated metabolite is 385.35, while that of the corresponding heptadeuterated metabolite is 392.39. (B) HPLC traces showing absorbance at 240 nm versus time for separation of crude apolar lipids extracted from HO1 cells grown on cholesterol and of authentic cholest-4-en-3-one as a reference. The retention time for the major peak was ~24.2 min. The traces for separation of crude apolar lipids extracted from WT and HO1 cells grown on heptadeuterated cholesterol are offset for clarity.
The m/z value of the 385.35 metabolite suggested that the 3-OH group was converted into a keto group because, in contrast to the hydroxyl group of cholesterol, it was not readily eliminated as a molecule of water during ionization. Thus, the metabolite was most likely cholest-5-en-3-one and/or cholest-4-en-3-one. In order to differentiate these possibilities, we isolated the cholesterol-derived metabolite by resolving the apolar lipid fraction extracted from M. tuberculosis cells exposed to cholesterol by reverse-phase HPLC. The conjugated ketone present in cholest-4-en-3-one has significant absorbance at 240 nm, facilitating detection of this candidate, while the presence of cholest-5-en-3-one was monitored at 210 nm. A single metabolite absorbing at 240 nm and co-eluting with authentic cholest-4-en-3-one was obtained in all tested extracts (Fig. 3B). Notably, the amount of the metabolite measured in each extract matched very well with what we observed by mass spectrometry. The corresponding chromatographic fractions were collected, extracted, and the identity of the metabolite was confirmed by LTQFT/MS and UV-vis spectroscopic comparisons with authentic cholest-4-en-3-one (Fig. S1). Thus, the substantial cellular accumulation of cholest-4-en-3-one in HO1 cells suggests that this compound is a physiological substrate for CYP125A1.
CYP125A1 binds and catalyzes the monooxygenation of cholest-4-en-3-one at C-27
Recombinant CYP125A1 was expressed in E. coli and the spectral properties of the purified protein were as reported previously (Capyk et al., 2009, McLean et al., 2009, Ouellet et al., 2009). CYP125A1 displayed the spectral properties of a ferric P450 with most of the heme iron (~70%) in a high spin state with a Soret band at 390 nm (Fig. 4A). The binding of a substrate-like molecule to P450 enzymes typically results in a type I-shift (transition from low spin to high spin) of the Soret band, as a result of the displacement of the water molecule coordinated to the heme iron. Cholest-4-en-3-one prepared in ethanol caused significant type-I spectral changes upon addition to CYP125A1. To solubilize the more hydrophobic cholesterol, 10% (w/v) β-methyl cyclodextrin (MBCD) was used. When both cholest-4-en-3-one and cholesterol stock solutions at 5 mM were prepared in 10% (w/v) MBCD, the addition of cholest-4-en-3-one to CYP125A1 resulted in the virtually complete conversion to the high-spin form, while a partial conversion was recorded with cholesterol (Fig. 4A). The KD values for both steroids were obtained from the spectral titration curves (Fig. 4B and 4C). The titration plots werebest fitted to a quadratic equation for tight binding (Equation 1, see Experimental Procedures) with KD values of 1.18 ± 0.11 and 0.107 ± 0.064 μM for cholest-4-en-3-one and cholesterol, respectively.
Fig. 4.

Binding of cholest-4-en-3-one and cholesterol to recombinant CYP125A1. (A) Absolute Soret region absorption spectrum of CYP125A1 protein (12.95 μM heme) in its resting (thin line), cholest-4-en-3-one (dashed line) cholesterol-bound (thick line) forms. The concentration dependence of ligand binding was deduced from the difference absorption changes obtained from the titration of protein (10 μM P450) with increasing concentrations of cholest-4-en-3-one (B) and cholesterol (C). Representative sets of the difference spectra that were recorded are shown in the insets. Spectrophotometric titrations were carried out as described in the Experimental Procedures section.
The enzymatic activity was reconstituted in vitro using the heterologous electron donor partners, spinach ferredoxin and ferredoxin reductase, and an NADPH-regenerating system. We observed the oxidation of both cholest-4-en-3-one and cholesterol after 20 min of incubation with CYP125A1, as judged by the appearance of a new peak in the GC-chromatogram (Fig. 5). The relative retention times and mass spectra were consistent with production of the C-27 hydroxylated forms of cholest-4-en-3-one and cholesterol. The assignment of the products was based on comparison with authentic samples, which exhibit characteristic mass spectra with diagnostic peaks (Fig. S3 and S4). At a substrate concentration of 50 μM, the rates of catalytic conversion of cholest-4-en-3-one and cholesterol were 9.2 and 1.95 nmol min−1 nmol−1 CYP125A1, respectively. Addition of the non-ionic detergent Tween-20 was previously shown to increase the enzymatic activity of the human P450 isoform CYP7A1 toward cholesterol (Mast et al., 2005). When Tween-20 was substituted for MBCD, we observed comparable CYP125A1 activity for cholest-4-en-3-one, but no activity with cholesterol. Moreover, the presence of Tween-20 resulted in the appearance of three new peaks in the GC-chromatogram (Fig. 5, bottom panel). The relative retention times and mass spectra were consistent with production of the 27-hydroxycholest-4-en-3-one and subsequent oxidation to the cholest-4-en-3-one-27-oic acid via the aldehyde cholest-4-en-3-one-27-al. The assignment of the two other oxidation products was based on analysis of their mass spectra, which exhibit diagnostic peaks (Fig. S5–S7).
Fig. 5.

Oxidation of cholest-4-en-3-one and cholesterol by CYP125A1. Offset GC traces for incubation of cholest-4-en-3-one (top and bottom panels) or cholesterol (middle panel) with CYP125A1 in reconstituted enzymatic activity assays. For the top and middle panels, the reactions were performed in the presence of 0.45% (w/v) MBCD, while that of the bottom panel was performed in the presence of 0.05% (v/v) Tween-20. The peak labeled S corresponds to the substrate cholest-4-en-3-one or cholesterol. The peaks labeled -CH2OH, –CHO and -COOH correspond to the products 27-hydroxy cholest-4-en-3-one, 27-al cholest-4-en-3-one TMS ethers, and 27-carboxylic acid cholest-4-en-3-one TMS ester (after derivatization for GC-MS), respectively. The inset of the bottom panel is a ~5x enlargement of product peaks region.
Overall structure of cholest-4-en-3-one-bound CYP125A1
The x-ray structure of the CYP125A1-cholest-4-en-3-one complex was determined to a resolution of 1.58 Å. Electron density for the substrate is unambiguously defined revealing a tightly shaped binding tunnel (Fig. 6) with fifteen residues surrounding cholest-4-en-3-one within 4 Å: F100, I104, D108, Q112, V115, L117, M200, P213, K214, S217, V263, V267, A268, T272 and W414 (Fig. 7). An additional eleven residues, I57, I109, V111, N118, T201, N203, D212, F260, M264, V313 and F316 are within 6 Å. Cholest-4-en-3-one aligns in the tunnel with the aliphatic side chain facing the distal surface of the heme prosthetic group and the 3-keto group pointing toward the protein surface (Fig. 6). The tunnel narrows down towards the catalytic site to tightly enclose the aliphatic side chain of cholest-4-en-3-one. Overall, the tunnel is extremely narrow in one dimension (Fig. 6A) but allows more space in the plane of the tetracycle (Fig. 6B). Binding of cholest-4-en-3-one caused conformational changes which are largely due to repositioning of the N-terminal portion of the I-helix and the H-helix to enclose the substrate in the active site. Upon substrate binding, the N-terminal portion of the I-helix bends to make hydrophobic contacts with the cholest-4-en-3-one molecule. The conformational adjustments resulted in an r.m.s.d of 0.78 Å between the ligand-free and ligand-bound structures.
Fig. 6.

Substrate binding tunnel. View of the cholest-4-en-3-one-bound CYP125A1 clipped by a plane through the substrate binding tunnel orthogonal (A) or parallel (B) to the plane of the tetracyclic sterol nucleus. Hydrophobic areas are orange, hydrophilic areas are blue. The residues preventing cholest-4-en-3-one from sliding toward the heme are labelled. The cholest-4-en-3-one color scheme is carbon grey, oxygen red. The image was generated using CHIMERA (Pettersen et al., 2004).
Fig. 7.

Cholest-4-en-3-one binding. Stereoscopic view of cholest-4-en-3-one (highlighted in yellow) surrounded by the CYP125A1 side chains (cyan) within 4 Å. Main chain atoms are shown for a couple of consecutive residues P213-K214 constituting a crevice where the sterol A-ring resides. 2Fo-Fc electron density map (grey mesh) was generated with the cholest-4-en-3-one coordinates omitted. For clarity, heme, Q112 and V115 are excluded from map calculation. Oxygen is coloured red, nitrogen blue, heme orange. The image was generated using PYMOL (DeLano, 2002).
Cholest-4-en-3-one binding in the active site
Two terminal methyl groups of the aliphatic chain bind at 4.30 Å and 4.83 Å from the Fe center with the methyl-branched fork pointing toward L117 and F316. The tertiary C-H bond at C-25 points toward A268 and is virtually parallel to the heme macrocycle, with a distance of 3.8 Å between C-25 and the Cα of A268. The unfavorable orientation of this bond disfavors hydroxylation despite the fact that the electron-rich nature of the tertiary C-H bond would normally make it a more reactive target for selective oxidation. Sliding of cholest-4-en-3-one along the tunnel to possibly alleviate this steric constraint is prevented by M200 and V267, which protrude between the van der Waals space of the axially oriented C-18 and C-19 methyl groups (Fig. 6A). On the other side of the tunnel, W414 fits into the corner between the sterol D-ring and the aliphatic chain, virtually locking cholest-4-en-3-one into the observed position (Fig. 6B).
Upon positioning of the aliphatic chain for hydroxylation, the keto group of cholest-4-en-3-one resides at the more hydrophilic end of the substrate binding tunnel surrounded by the residues D108, K214, N203 and D212. The keto group does not form direct H-bonding contacts with the protein side chains but participates, via water molecules, in an H-bonding network involving D108, K114, N203, D212 and S217. Given similar position of the hydroxyl group of cholesterol, we assume that it would interact with the same set of the residues. Two residues on the protein surface, K214 and D108, form a salt-bridge to isolate the substrate binding tunnel from the bulk solvent (Fig. 6).
Cholest-4-en-3-one is toxic for M. tuberculosis strains
A toxicity associated with cholesterol metabolism was observed when Δigr cells were incubated in the presence of two sources of carbon, glycerol and cholesterol (Chang et al., 2009). In order to specifically address the potential toxicity of cholest-4-en-3-one, cells were grown in minimal media containing cholest-4-en-3-one at two concentrations and glycerol as a second source of carbon. The growth was monitored by measuring the OD600 and a representative set of data is presented in Fig 8. When tested at a concentration of 0.01 mM (3.84 μg/ml), cholest-4-en-3-one completely blocked the growth of the HO1 cells, whereas the wild-type CDC1551 and H37Rv strains grew normally, and a lag in the growth was observed for HO11 (Fig. 5A). In order to test the possibility that the partial complementation in HO11 is due to the additive contributions from the five other genes of the igr locus, we transformed HO1 with a construct bearing the whole igr operon (HO12 strain). To confirm the specific role of CYP125A1 we also introduced in the chromosome a copy of the igr operon containing a mutation in cyp125A1 that replaces the essential heme axial cysteine ligand by an alanine (HO13). As shown in Fig. 8, HO12 showed partial complementation, whereas HO13, just like HO1 did not grow. This observation rules out a polarity effect on the transcription of the five other genes, and thus emphasizes the importance of CYP125A1 in the detoxification process regardless of the possible involvement of the other genes. It is still unclear why the complementation is partial, but we can hypothesize a non-optimal gene expression from the integrated constructs. In contrast, at a concentration of 0.1 mM (38.4 μg/ml), none of the CDC1551 strains grew, demonstrating that cholest-4-en-3-one can be toxic for M. tuberculosis. However, it appears that the degree of sensitivity varies among the wild-type isolates, as the lab strain H37Rv is more resistant, suggesting that it possesses compensatory steroid degrading activities.
Fig. 8.

Growth inhibition by cholest-4-en-3-one. Representative growth curves on minimal media with 0.1% (v/v) glycerol in combination with 0.01 mM cholest-4-en-3-one (top panels) or 0.1 mM cholest-4-en-3-one (bottom panels). The growth was monitored by measuring the absorbance at 600 nm in function of time.
Discussion
In this work, we used genetic, lipidomic, biochemical, and structural techniques to characterize the molecular function and physiological role of CYP125A1. The binding studies and the reconstitution of the enzymatic activity with recombinant CYP125A1 demonstrated that both cholest-4-en-3-one and cholesterol are substrates in vitro. We determined the first crystal structure of a CYP125A1 enzyme in complex with a physiological substrate. The study also led to the identification of an endogenously produced metabolite, cholest-4-en-3-one, that has toxic properties in M. tuberculosis.
Here, we not only extend the growing body of evidence that M. tuberculosis can utilize cholesterol as a source of carbon and energy (Brzostek et al., 2009, Pandey and Sassetti, 2008, Yam et al., 2009, Capyk et al., 2009, Hu et al. 2010, Nesbitt et al., 2009), but also provide new biochemical and structural evidence that confirm the key role of CYP125A1 in this process. First, the Δcyp125A1 mutant failed to grow in minimal media containing cholesterol. Second, the deletion mutant lost its ability to convert the side chain of cholesterol into the propionyl-CoA that is required for de novo synthesis of methyl-branched-containing PDIMs. Third, in vitro reconstitution of the enzymatic activity revealed that CYP125A1 efficiently catalyzes the C-27 hydroxylation of cholest-4-en-3-one and cholesterol and subsequently oxidizes the alcohol of the former to the carboxylic acid via the aldehyde intermediate. Based on other studies on R. jostii RHA1 (Rosloniec et al., 2009) and M. bovis BCG (Capyk et al., 2009), these CYP125A1 activities are required to initiate the degradation of the alkyl side chain, an obligatory step that precedes the degradation of the ring nucleus in M. tuberculosis.
Chang et al. reported that the H37Rv Δigr mutant strain is not capable of growth in a medium containing both glycerol and cholesterol, but still possesses detectable cholesterol side chain-degrading activity, as revealed by the incorporation of 14C atoms derived from the side chain into cellular lipids (Chang et al., 2009). A very recent study also suggested the existence of redundant cholesterol side chain degrading activities in M. tuberculosis (Capyk et al., 2009), as evidenced by the lack of effect of cyp125A1 inactivation in laboratory strain H37Rv for growth on cholesterol. As the same deletion performed in Mycobacterium bovis BCG resulted in the absence of growth on cholesterol, the authors proposed that the M. tuberculosis H37Rv genome encodes three additional enzymes potentially capable of initiating cholesterol side chain degradation. Two of these genes encode the cytochrome P450 enzymes CYP130A1 and CYP141A1. We addressed this possibility by attempting multiple times, without success, to detect the formation of any oxidation products from cholesterol or cholest-4-en-3-one catalyzed by these two P450 enzymes (data not shown). We conclude that at least these two enzymes are not involved in an alternative pathway of cholesterol metabolism. Although, the reported data suggest that compensatory activities may contribute to the overall catabolism of cholesterol in mycobacteria, the major catalyst in the oxidation of the steroid side chain in the physiologically relevant clinical isolate CDC1551 is undoubtedly CYP125A1.
Two instances of toxicity associated with cholesterol metabolism in M. tuberculosis cells were recently reported (Chang et al., 2009, Yam et al., 2009). HsaC, an iron-dependent extradiol dioxygenase that cleaves catechols, causes a loss of viability in the ΔhsaC mutant, consistent with catechol toxicity (Yam et al., 2009). In the second case, deletion of the igr operon resulted in a cell poisoning during in vitro growth in liquid culture in the presence of cholesterol and a significant lag in M. tuberculosis dissemination during the initial phase of mouse infection (Chang et al., 2009). The toxic effect was attributed by the authors to the accumulation of an unidentified, cholesterol-derived metabolite even when the cells were grown in the presence of an additional carbon source (e.g. glycerol). Indeed, the inactivation of the mce4 locus, encoding for a steroid transporter (Mohn et al., 2008), in the Δigr strain abolished the toxicity associated with cholesterol and resulted in attenuation of virulence during the persistent phase of mouse infection. Here, we show that specific deletion of the cyp125A1 component of the igr operon results in an inhibitory effect due to the accumulation of cholest-4-en-3-one. Interestingly, deletion of the fadA5 gene that encodes ketoacyl-coenzyme A thiolase does not give rise to any toxicity effect even though it is essential for cholesterol side chain degradation during the persistent phase of infection (Nesbitt et al., 2009). This observation butresses our proposal that cholest-4-en-3-one, and possibly its side chain oxidized products, are responsible for the inhibitory effect. Altogether, these observations strongly suggest that the biological function of CYP125A1, and potentially other proteins encoded by the igr operon (Chang et al., 2009), might not be restricted to the utilization of cholesterol during the chronic phase of infection, but might be also crucial during the initial phase to alleviate the toxicity associated with cholesterol catabolism.
The interaction mode of CYP125A1 with a substrate was crystallographically defined in this work by the structure of the CYP125A1-cholest-4-en-3-one complex. This x-ray structure validates the accuracy of the cholesterol binding mode previously modeled by McLean et al. (2009). The hydrophobic substrate binding tunnel narrows down towards the catalytic site to tightly enclose the aliphatic side chain of cholest-4-en-3-one. The tetracyclic steroid nucleus of the co-crystallized cholest-4-en-3-one and that of the modeled cholesterol reside differently within the active site than that of androstenedione in its crystal structure with CYP125 (McLean et al., 2009), suggesting that the aliphatic side chain plays a dominant role in establishing the catalytically productive substrate orientation
Despite the electronic factor favoring hydroxylation of the tertiary C-H bond, disposition of the methyl carbons of the methyl-branched aliphatic chain of cholest-4-en-3-one in the CYP125A1 active site favors oxidation of a primary C-H bond of the C-26/C-27 methyl groups with a slight preference for C-27 and thus for formation of the S-stereoisomer from the prochiral center. This conclusion is similar to that drawn from the CYP124-phytanic acid complex (Johnston et al., 2009). For both enzymes, steric constraints rather than the differential electronic properties of the C-H bonds dictate the regioselectivity of the reaction.
The differences between cholest-4-en-3-one and its precursor cholesterol are the planarity of the sterol A-ring and the electronic properties of the keto- versus hydroxyl-substituents at C-3. Both substituents are capable of participating in the H-bonding network, which makes it likely that the hydroxyl group of cholesterol interacts with the same set of residues as does the keto group of cholest-4-en-3-one. Conjugation between the carbonyl group and the adjacent double bond in cholest-4-en-3-one flattens the A-ring compared to the chair conformation of its saturated counterpart in cholesterol and creates a curvature in the cholest-4-en-3-one molecule. In the crystal structure, the A-ring of cholest-4-en-3-one resides in the protein surface crevice formed by the side chains of the FG-loop residues P213 and K214 (Fig 7). The well-established mobility of this region in cytochromes P450 apparently allows the protein to adjust its conformation to accommodate cholesterol.
Both the physiological importance of the cholesterol degrading pathway and the toxic effect of the cholest-4-en-3-one intermediate raise the possibility of targeting CYP125A1 for therapeutic purposes. The crystal structure of the CYP125A1-cholest-4-en-3-one complex determined in this work defines in high resolution the active site of the enzyme and provides a platform for designing inhibitors with potential to target tuberculosis. Furthermore, the identification of cholest-4-en-3-one as a toxic agent in M. tuberculosis opens up the possibility that identification of the site of action of this molecule may reveal new targets for antimycobacterial drug design.
Experimental Procedures
Materials
Cholest-4-en-3-one, cholesterol, spinach ferredoxin, spinach ferredoxin-NADP+-reductase, bovine liver catalase, glucose-6-phosphate dehydrogenase, glucose-6-phosphate and β-methyl cyclodextrin were purchased from Sigma-Aldrich (St. Louis, MO). Heptadeuterated cholesterol ([25, 26, 26, 26, 27, 27, 27-D7]-cholesterol), was obtained from Avanti Polar Lipids (Alabaster, AL). 4-cholesten-3-one-(25S)-carboxylic acid was obtained from Steraloids (Newport, RI).
Strains, media and culture conditions
M. tuberculosis CDC1551 cells were cultured in either 7H9 medium supplemented with 10% of oleic acid-albumin-dextrose complex (OADC), 0.2% glycerol and 0.05% Tween-80 or on 7H 10 solid agar medium with the same supplements excluding Tween-80, plus cycloheximide (50 mg l−1). Hygromycin and zeocin (50 mg l−1) were used where necessary. For growth on cholesterol log-phase cells grown in 7H9 medium cells were subcultured in a defined minimal medium (1 g l−1 KH2PO4, 2.5 g l−1 Na2HPO4, 0.5 g l−1 asparagine, 50 mg l−1 ferric ammonium citrate, 10 mg l−1 MgSO4·7H2O, 0.5 mg l−1 CaCl2 0.1 mg l−1 ZnSO4). For growth in the presence of cholest-4-en-3-one, glycerol was also included at a concentration of 0.1% (v/v). Cholesterol and cholest-4-en-3-one were dissolved at a concentration of 200 mM in tyloxapol/ethanol (1:1) and warmed at 65 °C for 30 min before addition. Equal volumes of diluted cell suspension and 0.2 mM steroid-containing medium were mixed to obtain a final concentrationof 0.1 mM steroids at an OD600 of 0.05.
Construction of the cyp125A1 disrupted mutant by specialized transduction in M. tuberculosis CDC1551
The Δcyp125A1 mutant strain was created by homologous recombination using specialized transducing phages, as described (Bardarov et al., 2002). Briefly, a substrate for allelic exchange at cyp125A1 was constructed by amplifying the flanking regions of cyp125A1 by PCR from M. tuberculosis genomic DNA using the primers listed in Table S1. Flanking regions of cyp125A1 802 bp 5′ flank beginning at nucleotide 33 of cyp125A1 and 817 bp beginning at nucleotide 1002 were amplified, cloned with the TA cloning kit (Invitrogen), and sequenced. The two DNA fragments were cloned successively into pJSC407 (Grundner et al., 2008) on each side of a hygromycin cassette flanked by resolvase sites. The specialized transduction phage was incubated with concentrated wild-type CDC1551 M. tuberculosis cells for 4 h at 37 °C. Cells were then plated on 7H10 plates containing hygromycin. After 3 weeks, colonies were picked and screened for the deletion by Southern blotting using the 5′ flanking region of cyp125A1 as a probe. The resulting deletion replaced 969 bp of cyp125A1 (amino acids 11-334), leaving the 5′ flanking region of the gene fadE28 gene (Fig. 1) intact.
Complementation of the cyp125A1 disrupted mutant
The coding sequences of cyp125A1 were amplified from M. tuberculosis genomic DNA by using Pfu Turbo polymerase (Stratagene) and the primers listed in Table S1. To confirm the DNA sequence, the PCR fragment was first cloned into a pCR-BluntII-TOPO vector (Invitrogen) and then the NdeI-XbaI digested fragment was subcloned downstream of the constitutive hsp60 promoter into a derivative of the integrative vector pMV361 (Hickey et al., 1996) that confers resistance to zeocin to give pHO11. The complete igr operon was also amplified by PCR using the robust and high-fidelity polymerase Phusion (New England BioLabs) and the primers listed in Table S1, according to the protocol recommended by the manufacturer for a long template. The amplified fragment was first cloned in pCR-BluntII-TOPO vector for sequencing. The KpnI digest fragment was then subcloned into a derivative of the integrative vector pMV306 (Lee et al., 1991) that confers resistance to zeocin to give pHO12. The cysteine found at the position 377 in CYP125A1 was changed to an alanine by site-directed mutagenesis (QuikChange site-directed mutagenesis kit, Stratagene) using the igr operon as a template to give pHO13. The three constructs were electroporated into Δcyp125A1 cells, and transformants were isolated at 37 °C on 7H10 plates containing 50 μg/ml zeocin.
Extraction of lipids
For lipidomic experiments, cells were precultured in 7H9 medium, washed in the same medium lacking glycerol, synchronized to an OD600 of 0.2, and grown for an additional 24 h in the presence of 0.1% (v/v) glycerol or 0.1 mM cholesterol. Cells (25 ml) were harvested by centrifugation, washed twice with 25 ml of 15% isopropanol in saline and resuspended in saline at a similar OD600 for all strains. Samples (1 ml) were extracted with 5 ml of chloroform/methanol (17:1) for 2 h. The lower organic phase containing apolar lipids was collected. The remaining aqueous sample material was reextracted with 5 ml chloroform/methanol (2:1) for 2 h to extract the polar lipid fraction. The lipid extracts were evaporated under a stream of argon and resuspended in 1.5 ml of methanol. Tween-80 was removed from the lipid solutions by precipitation using a cobalt/thiocyanate solution and back-extraction with hexane, according to a published procedure (Shen et al., 2004). Finally, the lipid extracts were dissolved in 1 ml of chloroform/methanol (2:1).
Mass spectrometry
All data were acquired on an LTQFT/MS instrument (ThermoFisher Scientific, Bremen, Germany). Lipid samples were diluted 10-fold in chloroform:methanol:isopropanol (1:2:4) containing 7.5 mM ammonium acetate before being introduced into the mass spectrometer though a static (medium) nanospray tip (Proxeon, Odense, Denmark). Electrospray voltages were 1000 volts for positive and −700 volts for negative ionization. For molecular weight measurement, 10–100 scans were collected and averaged to produce the final spectra, with mass resolution of 100,000 and 25,000 for lipid and cholesterol samples, respectively. The final spectra for multi-stage MSMS (up to four stages) were acquired in the FT mode.
Isolation and identification of the cholesterol-derived metabolites from crude lipid extracts
Analytical HPLC was used to isolate cholesterol-derived metabolites accumulating inΔcyp125A1 cells. The HPLC system was an Agilent Technologies 1200 Series equipped with a photodiode array detector connected to Polaris C8 column (250 × 4.6 mm i.d. stainless steel, 5 μm particles, Varian). The constituents of lipid extracts were separated using an isocratic mobile phase consisting of water:acetonitrile:methanol (25:25:50) with 0.1% (v/v) formic acid at a flow rate of 1 ml min−1. The UV detector wavelength was set at 210 and 240 nm for detection of cholest-5-en-3-one and cholest-4-ene-3-one derivatives, respectively.
Cloning, Overexpression and Purification of CYP125A1
The cyp125A1 gene was cloned from M. tuberculosis H37Rv genomic DNA as described previously (Ouellet et al., 2009). Protein was expressed in E. coli DH5α cells co-transformed with pCWori/cyp125 and pGro7 plasmids, encoding for CYP125A1 and GroEL/ES chaperones, respectively. Cells were grown at 37 °C in Terrific broth (TB) medium containing 100 μg ml−1 of ampicillin, 40 μg ml−1 of chloramphenicol and 0.2% (w/v) of L-arabinose. When OD590 = 0.8 expression of CYP125A1 was induced with 500 μM Isopropyl β-D-1-thiogalactopyranoside (IPTG) in the presence of 0.25 mM δ-aminolevulinic acid (δ-ALA). The temperature was decreased from 37 °C to 25 °C and the culture continued for 36 h. The protein was purified as described previously (Ouellet et al., 2008). The protein preparation used in this study exhibited a typical P450 CO-deoxy difference spectrum (Fig. S2) and contained a significant amount of P420. The actual concentration of P450 was determined from difference spectra using the extinction coefficient ε450–490 = 91000 M−1 cm−1 (Omura and Sato, 1964). The total heme b content was quantified by the pyridine hemochrome method (Appleby, 1978).
Spectroscopic binding assays
All ligand binding assays were performed by spectrophotometric titration in 50 mM potassium phosphate buffer (pH 7.4) containing 0.1 mM EDTA using a Cary UV-visible scanning spectrophotometer (Varian). Stock solutions of steroids at 5 mM were prepared in 10% (w/v) β-methyl cyclodextrin (BMCD) in buffer. To account for the absorbance of the tested compounds, 1 ml of protein (10 μM) in buffer was placed in the first chamber of two split cuvettes and 1 ml of buffer was placed in the second chamber. After background scanning, equal volumes (0.25 to 1.0 μl) of ligand solution were titrated into both the second chamber of the reference cuvette containing only buffer and the first chamber containing the protein. The same volume of BMCD was added into the alternate chambers to correct for solvent effects. Difference spectra were recorded from 300 to 500 nm. To determine the KD values, titration data points were fitted to the quadratic equation using the Kaleidagraph software (Synergy). In this equation, Aobs is the absorption shift determined at any ligand concentration; Amax is the maximal absorption shift obtained at saturation; KD is the apparent dissociation constant for the inhibitor-enzyme complex; [Et] is the total enzyme concentrationused; [S] is the ligand concentration.
| Equation 1 |
CYP125A1 in vitro activity assay
CYP125A1 reactions (0.5 ml) were carried out in glass tubes at 25 °C using 50 mM potassium phosphate, pH 7.5 containing 0.05% (v/v) Tween-20 or 0.45% (w/v) BCMD. CYP125A1 (1.0μM) was pre-incubated for 2 h at 25 °C with substrate. Reactions were initiated by adding spinach ferredoxin, spinach ferredoxin-NADP+ reductase, catalase, magnesium chloride and an NADPH-regenerating system consisting of glucose-6-phosphate dehydrogenase and glucose-6-phosphate as described elsewhere (Johnston et al., 2009). The CYP125A1 specific activity values were determined at saturating substrate concentrations. The enzymatic reactions or negative controls were quenched by adding an equal volume of 1N HCl. Substrates and reaction products were extracted, derivatized and analyzed by GC/MS, as described previously (Johnston et al., 2009).
Crystallization, data collection and crystal structure determination
The initial screening of crystallization conditions was performed by using commercial high-throughput screening kits (Hampton Research), a nanoliter drop-setting Mosquito robot (TTP; Labtech) operating with 96-well plates, and a hanging-drop crystallization protocol. The protein was from a 1 mM frozen stock in 20 mM Tris (pH 7.5), 0.5 mM EDTA and 0.25M NaCl. Prior to crystallization the protein was diluted to 0.2 mM by mixing with 10 mM Tris HCl (pH 7.5) supplemented with 1 mM cholest-4-en-3-one. Crystals of the CYP125-cholest-4-en-3-one complex grew at 4 °C from 0.2 M ammonium sulfate, 0.1 M bis-Tris, pH 5.5, and 25% PEG 3350. Crystals from the 200-nanolitre Mosquito drops were used for diffraction data collection. To harvest, the crystals were cryo-protected by plunging them into a drop of reservoir solution supplemented with 20% (v/v) glycerol and flash frozen in liquid nitrogen. Diffraction data were collected at 100–110 K at beamline 8.3.1, Advanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA. Data indexing, integration and scaling were conducted by using ELVES automated software suite (Holton and Alber, 2004). Crystal structures were determined by molecular replacement using the atomic coordinates of CYP125A1 (PDB ID CODE: 3IVY) as a search model. Automated model building using ARP/wARP (Langer et al., 2008) placed the majority of the polypeptide chain in the asymmetric unit. The remaining residues were built manually with COOT (Emsley and Cowtan, 2004), alternated with positional refinement using REFMAC5 (Murshudov et al., 1997, Winn et al., 2001). Data collection and refinement statistics are provided in Table 1.
Table 1.
Data collection and refinement statistics.
| Protein | CYP125A1 |
|---|---|
| Ligand | cholest-4-en-3-one |
| PDB ID | 2×5W |
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 73.7, 76.0, 79.6 |
| Molecules in AU | 1 |
| Wavelength | 1.1159 |
| Resolution (Å) | 1.58 |
| Rsym or Rmerge (%) | 6.7 (58.3) |
| I/σI | 10.7 (1.6) |
| Completeness (%) | 99.3 (95.6) |
| Redundancy | 4.4 (2.8) |
| Refinement | |
| No. reflections | 58287 |
| Rwork/Rfree (%) | 15.3/20.0 |
| No. atoms | |
| Protein | 3311 |
| Heme | 43 |
| Ligand | 28 |
| Water | 438 |
| Mean B value | 20.3 |
| B-factors | |
| Protein | 19.2 |
| Heme | 10.5 |
| Ligand | 14.1 |
| Water | 30.8 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.025 |
| Bond angles (°) | 2.0 |
Values in parentheses are for highest-resolution shell.
Supplementary Material
Acknowledgments
We thank Dr. Chiung-Kuang Chen and the staff members of beamline 8.3.1, James Holton, George Meigs and Jane Tanamachi, at the Advanced Light Source at Lawrence Berkeley National Laboratory, for assistance with data collection. This work was supported by National Institutes of Health RO1 Grants GM25515 (to P.O.M.), AI74824 (to P.O.M.), AI51667 (to J.S.C.), GM078553 (to L.M.P.) and NIH NCRR grants RR01614 and RR019934 (to A.L.B). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Data deposition note: The atomic coordinates and structure factors have been deposited in the Protein Data Bank (PDB ID code 2X5W), Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
References
- (WHO), W. H. O. World Health Organization Facts Sheets on Tuberculosis 2009 [Google Scholar]
- Appleby CA. Purification of Rhizobium cytochromes P-450. Methods Enzymol. 1978;52:157–166. doi: 10.1016/s0076-6879(78)52018-1. [DOI] [PubMed] [Google Scholar]
- Bardarov S, Bardarov S, Jr, Pavelka MS, Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR., Jr Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology. 2002;148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- Boshoff HI, Barry CE., 3rd A low-carb diet for a high-octane pathogen. Nat Med. 2005a;11:599–600. doi: 10.1038/nm0605-599. [DOI] [PubMed] [Google Scholar]
- Boshoff HI, Barry CE., 3rd Tuberculosis - metabolism and respiration in the absence of growth. Nat Rev Microbiol. 2005b;3:70–80. doi: 10.1038/nrmicro1065. [DOI] [PubMed] [Google Scholar]
- Brzostek A, Dziadek B, Rumijowska-Galewicz A, Pawelczyk J, Dziadek J. Cholesterol oxidase is required for virulence of Mycobacterium tuberculosis. FEMS Microbiol Lett. 2007;275:106–112. doi: 10.1111/j.1574-6968.2007.00865.x. [DOI] [PubMed] [Google Scholar]
- Brzostek A, Pawelczyk J, Rumijowska-Galewicz A, Dziadek B, Dziadek J. Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J Bacteriol. 2009;191:6584–6591. doi: 10.1128/JB.00488-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capyk JK, Kalscheuer R, Stewart GR, Liu J, Kwon H, Zhao R, et al. Mycobacterial cytochrome P450 125 (cyp125) catalyzes the terminal hydroxylation of c27 steroids. J Biol Chem. 2009;284:35534–35542. doi: 10.1074/jbc.M109.072132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JC, Harik NS, Liao RP, Sherman DR. Identification of mycobacterial genes that alter growth and pathology in macrophages and in mice. J Infect Dis. 2007;196:788–795. doi: 10.1086/520089. [DOI] [PubMed] [Google Scholar]
- Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, Sherman DR. igr Genes and Mycobacterium tuberculosis cholesterol metabolism. J Bacteriol. 2009;191:5232–5239. doi: 10.1128/JB.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The Pymol molecular graphics system. San Carlos, CA, USA: DeLano Scientific; 2002. [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fleischmann RD, Alland D, Eisen JA, Carpenter L, White O, Peterson J, et al. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol. 2002;184:5479–5490. doi: 10.1128/JB.184.19.5479-5490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundner C, Cox JS, Alber T. Protein tyrosine phosphatase PtpA is not required for Mycobacterium tuberculosis growth in mice. FEMS Microbiol Lett. 2008;287:181–184. doi: 10.1111/j.1574-6968.2008.01309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MJ, Arain TM, Shawar RM, Humble DJ, Langhorne MH, Morgenroth JN, Stover CK. Luciferase in vivo expression technology: use of recombinant mycobacterial reporter strains to evaluate antimycobacterial activity in mice. Antimicrob Agents Chemother. 1996;40:400–407. doi: 10.1128/aac.40.2.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holton J, Alber T. Automated protein crystal structure determination using ELVES. Proc Natl Acad Sci USA. 2004;101:1537–1542. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, van der Geize R, Besra GS, Gurcha SS, Liu A, Rohde M, Singh M, Coates A. 3-Ketosteroid 9alpha-hydroxylase is an essential factor in the pathogenesis of Mycobacterium tuberculosis. Mol Microbiol. 2010;75:107–121. doi: 10.1111/j.1365-2958.2009.06957.x. [DOI] [PubMed] [Google Scholar]
- Jacobs W, Brennan P, Curlin G, Ginsberg A, Adams M, Fleischmann R, et al. Comparative sequencing. Science. 1996;274:17–18. doi: 10.1126/science.274.5284.17a. [DOI] [PubMed] [Google Scholar]
- Jain M, Petzold CJ, Schelle MW, Leavell MD, Mougous JD, Bertozzi CR, et al. Lipidomics reveals control of Mycobacterium tuberculosis virulence lipids via metabolic coupling. Proc Natl Acad Sci USA. 2007;104:5133–5138. doi: 10.1073/pnas.0610634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JB, Kells PM, Podust LM, Ortiz de Montellano PR. Biochemical and structural characterization of CYP124: a methyl-branched lipid omega-hydroxylase from Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2009;106:20687–20692. doi: 10.1073/pnas.0907398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Pascopella L, Jacobs WR, Jr, Hatfull GF. Site-specific integration of mycobacteriophage L5: integration-proficient vectors for Mycobacterium smegmatis, Mycobacterium tuberculosis, and bacille Calmette-Guerin. Proc Natl Acad Sci USA. 1991;88:3111–3115. doi: 10.1073/pnas.88.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca C, Tsenova L, Barry CE, 3rd, Bergtold A, Freeman S, Haslett PA, et al. Mycobacterium tuberculosis CDC1551 induces a more vigorous host response in vivo and in vitro, but is not more virulent than other clinical isolates. J Immunol. 1999;162:6740–6746. [PubMed] [Google Scholar]
- Mast N, Graham SE, Andersson U, Bjorkhem I, Hill C, Peterson J, Pikuleva IA. Cholesterol binding to cytochrome P450 7A1, a key enzyme in bile acid biosynthesis. Biochemistry. 2005;44:3259–3271. doi: 10.1021/bi047566a. [DOI] [PubMed] [Google Scholar]
- McLean KJ, Lafite P, Levy C, Cheesman MR, Mast N, Pikuleva IA, et al. The structure of Mycobacterium tuberculosis CYP125: Molecular basis for cholesterol binding in a P450 needed for host infection. J Biol Chem. 2009 doi: 10.1074/jbc.M109.032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn WW, van der Geize R, Stewart GR, Okamoto S, Liu J, Dijkhuizen L, Eltis LD. The actinobacterial mce4 locus encodes a steroid transporter. J Biol Chem. 2008;283:35368–35374. doi: 10.1074/jbc.M805496200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Elias EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005;11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nesbitt NM, Yang X, Fontan P, Kolesnikova I, Smith I, Sampson NS, Dubnau E. A thiolase of M. tuberculosis is required for virulence and for production of androstenedione and androstadienedione from cholesterol. Infect Immun. 2009 doi: 10.1128/IAI.00893-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes. II. Solubilization, purification, and properties. J Biol Chem. 1964;239:2379–2385. [PubMed] [Google Scholar]
- Ouellet H, Lang J, Couture M, Ortiz de Montellano PR. Reaction of Mycobacterium tuberculosis cytochrome P450 enzymes with nitric oxide. Biochemistry. 2009;48:863–872. doi: 10.1021/bi801595t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellet H, Podust LM, Ortiz de Montellano PR. Mycobacterium tuberculosis CYP130: crystal structure, biophysical characterization, and interactions with antifungal azole drugs. J Biol Chem. 2008;283:5069–5080. doi: 10.1074/jbc.M708734200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci USA. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Rengarajan J, Bloom BR, Rubin EJ. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci USA. 2005;102:8327–8332. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosloniec KZ, Wilbrink MH, Capyk JK, Mohn WW, Ostendorf M, van der Geize R, et al. Cytochrome P450 125 (CYP125) catalyses C26-hydroxylation to initiate sterol side-chain degradation in Rhodococcus jostii RHA1. Mol Microbiol. 2009;74:1031–1043. doi: 10.1111/j.1365-2958.2009.06915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, et al. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: Insights into the phagosomal environment. J Exp Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen CF, Hawari J, Kamen A. Micro-quantitation of lipids in serum-free cell culture media: a critical aspect is the minimization of interference from medium components and chemical reagents. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;810:119–127. doi: 10.1016/j.jchromb.2004.07.020. [DOI] [PubMed] [Google Scholar]
- Sherman DR, Voskuil M, Schnappinger D, Liao R, Harrell MI, Schoolnik GK. Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha-crystallin. Proc Natl Acad Sci USA. 2001;98:7534–7539. doi: 10.1073/pnas.121172498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valway SE, Sanchez MP, Shinnick TF, Orme I, Agerton T, Hoy D, et al. An outbreak involving extensive transmission of a virulent strain of Mycobacterium tuberculosis. N Engl J Med. 1998;338:633–639. doi: 10.1056/NEJM199803053381001. [DOI] [PubMed] [Google Scholar]
- Van der Geize R, Yam K, Heuser T, Wilbrink MH, Hara H, Anderton MC, et al. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci USA. 2007;104:1947–1952. doi: 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuil MI, Visconti KC, Schoolnik GK. Mycobacterium tuberculosis gene expression during adaptation to stationary phase and low-oxygen dormancy. Tuberculosis (Edinb) 2004;84:218–227. doi: 10.1016/j.tube.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- Yam KC, D’Angelo I, Kalscheuer R, Zhu H, Wang JX, Snieckus V, et al. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 2009;5:e1000344. doi: 10.1371/journal.ppat.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Nesbitt NM, Dubnau E, Smith I, Sampson NS. Cholesterol metabolism increases the metabolic pool of propionate in Mycobacterium tuberculosis. Biochemistry. 2009;48:3819–3821. doi: 10.1021/bi9005418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.