

Abstract
Copy number variation (CNV) of DNA sequences is functionally significant but has yet to be fully ascertained. We have constructed a first-generation CNV map of the human genome through the study of 270 individuals from four populations with ancestry in Europe, Africa or Asia (the HapMap collection). DNA from these individuals was screened for CNV using two complementary technologies: single nucleotide polymorphism (SNP) genotyping arrays, and clone-based comparative genomic hybridization. 1,447 copy number variable regions covering 360 megabases (12% of the genome) were identified in these populations; these CNV regions contained hundreds of genes, disease loci, functional elements and segmental duplications. Strikingly, these CNVs encompassed more nucleotide content per genome than SNPs, underscoring the importance of CNV in genetic diversity and evolution. The data obtained delineate linkage disequilibrium patterns for many CNVs, and reveal dramatic variation in copy number among populations. We also demonstrate the utility of this resource for genetic disease studies.
Introduction
Genetic variation in the human genome takes many forms, ranging from large microscopically-visible chromosome anomalies to single nucleotide changes. Recently, multiple studies have discovered an abundance of sub-microscopic copy number variation of DNA segments ranging from kilobases (kb) to megabases (Mb) in size 1-8. Deletions, insertions, duplications, and complex multi-site variants 9, collectively termed copy number variants (CNVs) or copy number polymorphisms (CNPs), are found in all humans 10 and other mammals examined 11. We defined a CNV as a DNA segment that is 1 kb or larger and present at variable copy number in comparison with a reference genome 10. A CNV can be simple in structure, such as tandem duplication, or may involve complex gains or losses of homologous sequences at multiple sites in the genome (Supplementary Figure 1).
An early association of CNV with a phenotype was described 70 years ago, with the duplication of the Bar gene in Drosophila melanogaster being shown to cause the Bar eye phenotype 12. CNVs influence gene expression, phenotypic variation and adaptation by disrupting genes and altering gene dosage 7,13-15, and can cause disease, as in microdeletion or microduplication disorders 16-18, or confer risk to complex disease traits such as HIV-1 infection and glomerulonephritis 19,20. CNVs often represent an appreciable minority of causative alleles at genes at which other types of mutation are strongly associated with specific diseases: CHARGE syndrome 21, Parkinson and Alzheimer disease 22,23. Furthermore, CNVs can influence gene expression indirectly through position effects, predispose to deleterious genetic changes, or provide substrates for chromosomal change in evolution 10,11,17,24.
In this study, we investigated genome-wide characteristics of CNV in four populations with different ancestry, and classified CNVs into different types according to their complexity and whether copies have been gained or lost (Supplementary Figure 1). To maximize the utility of these data and the potential for integration of CNVs with SNPs for genetic studies, we performed experiments with the International HapMap DNA and cell-line collection 25 derived from apparently healthy individuals. The result is the first comprehensive map of copy number variation in the human genome, which provides an important resource for studies of genome structure and human disease.
Two platforms for assessing genome-wide copy number variation
The HapMap collection comprises four populations: 30 parent-offspring trios of the Yoruba from Nigeria (YRI), 30 parent-offspring trios of European descent from Utah, USA (CEU), 45 unrelated Japanese from Tokyo, Japan (JPT) and 45 unrelated Han Chinese from Beijing, China (CHB). Genomic DNA from EBV-transformed lymphoblastoid cell-lines was used.
Two technology platforms were used to assess CNV (Figure 1): (i) comparative analysis of hybridization intensities on Affymetrix GeneChip® Human Mapping 500K Early Access Arrays (500K EA), in which 474,642 SNPs were analysed and (ii) comparative genomic hybridization with a Whole Genome TilePath (WGTP) array that comprises 26,574 large-insert clones representing 93.7% of the euchromatic portion of the human genome 26.
Figure 1. Protocol outline for two copy number variation (CNV) detection platforms.

The experimental procedures for Comparative Genome Hybridization (CGH) on the WGTP array and Comparative Intensity Analysis on the 500K EA platform are shown schematically (see Supplementary Methods for details), for a comparison of two male genomes (NA10851 and NA19007). The genome profile shows the log2 ratio of copy number in these two genomes chromosome-by-chromosome. The 500K EA data is smoothed over a 5-probe window. Below the genome profiles are zoomed plots of chromosome 8, and a 10Mb window containing a large duplication in NA19007 identified on both platforms (indicated by the red bracket).
Stringent quality control criteria were set for each platform and experiments were repeated for 82 individuals on the WGTP and 15 individuals on the 500K EA platform. The quality of the final datasets was assessed by the standard deviation among log2 ratios of autosomal probes (after normalisation and filtering for cell-line artefacts), which for the WGTP platform was 0.047 (Supplementary Figure 2), and for the 500K EA platform was 0.220, both of which are improvements on published data 8,27.
The different nature of the two datasets required the development of distinct algorithms to identify CNVs. In essence, these algorithms segment a continuous distribution of intensity ratios into discrete regions of copy number variation. To train the threshold parameters, we attempted to validate experimentally 203 CNVs that had been defined with varying degrees of confidence in two well-characterized genomes 4,5,7 (NA10851 and NA15510). By performing technical replicate experiments on both platforms we assessed the proportion of CNV calls that were false positives for different algorithm parameters across a set of experiments representing the spectrum of data quality. The threshold parameters for both algorithms were set to achieve an average false positive rate per experiment beneath 5% (Methods, Supplementary Methods and Supplementary Tables 1-4 26,28).
As all DNAs were derived from lymphoblastoid cell-lines, we differentiated somatic artefacts (such as culture-induced rearrangements and aneuploidies) from germline CNVs. We karyotyped all available 268 HapMap cell-lines (Supplementary Table 5) and sought evidence for chromosomal abnormalities in the WGTP and 500K EA intensity data. We identified 30 cell-lines with unusual chromosomal constitutions (Supplementary Table 5 and Supplementary Figure 3), and removed the aberrant chromosomes from further analyses. Chromosomes 9, 12 and X, seemed particularly prone to trisomy. For a cell-line with mosaic trisomy of chromosome 12, we confirmed by array-CGH that this trisomy was not apparent in blood DNA from the same individual (Supplementary Figure 4). Furthermore, we sought signals of somatic deletions within the SNP genotypes of HapMap trios. A somatic deletion in a parental genome manifests as a cluster of SNPs at which alleles present in the offspring are not found in either parent 5. We assessed all of our preliminary CNV calls in 120 trio parents and found that 17 (of 4,758) fell in genomic regions that harbour highly significant clusters of HapMap Phase II SNP genotypes compatible with a somatic deletion in a parental genome (Supplementary Table 5A, Supplementary Figure 5, Supplementary Note). These putative cell-line artefacts were removed from further analyses. Extrapolating this analysis to the entire HapMap collection, suggests that less than 0.5% of the deletions we observed were likely to have been somatic artefacts.
The quality of resultant CNV calls were assessed in additional ways 26,28. Technical replicate experiments (triplicates for ten individuals) demonstrated that CNV calls are highly replicable (Supplementary Table 6), and that noisier experiments are characterized by higher false negative rates, rather than higher false positive rates (Supplementary Figure 2). Heritability of CNVs within trios was investigated at 67 biallelic CNVs at which CNV genotypes could be inferred (Figure 2, Supplementary Table 7). Of 12,060 biallelic CNV genotypes, only ~0.2% exhibited Mendelian discordance, which likely reflects the genotyping error rate rather than the rate of de novo events at these loci. Additional locus-specific experimental validation was performed on subsets of CNVs (Supplementary Table 4). CNVs called in only a single individual (‘singleton CNVs’) are more likely to be false positives compared to CNVs identified in several individuals. We attempted to validate 50 singleton CNVs called on only one platform (25 from each platform), and 14 singleton CNVs called on both platforms. All 14 singleton CNVs replicated by both platforms were verified as true positives, while 38 out of 50 of CNVs called by only one platform were similarly confirmed (false positive rate of 24%). Extrapolating these validation rates across the entire dataset, suggests that only 8% (24% multiplied by the frequency of singleton CNVs called on only one platform) of the copy number variable regions we identify (see below) are likely to be false positives.
Figure 2. Heritability of 5 CNVs in 4 HapMap trios.
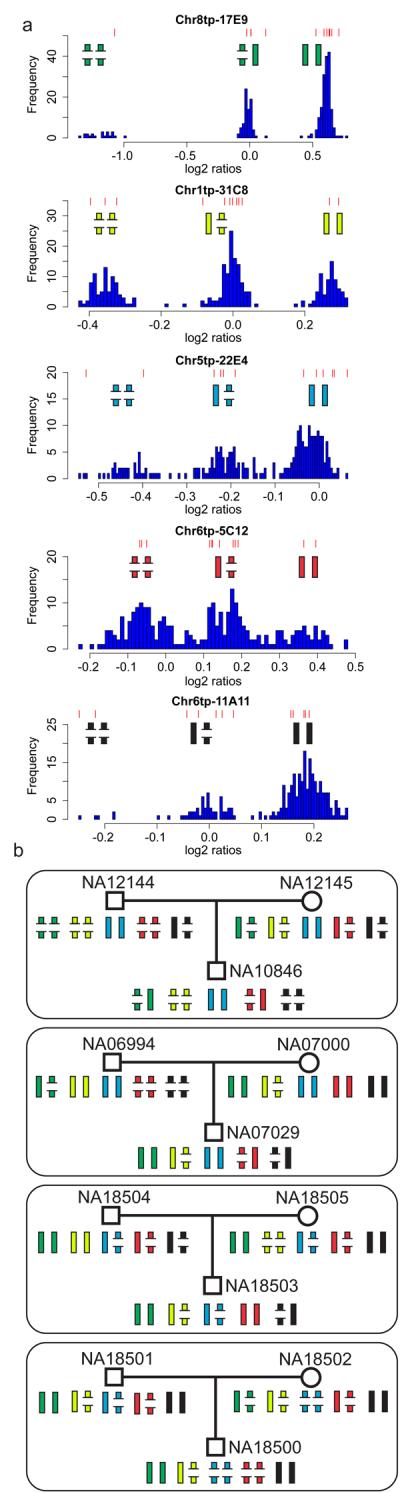
Panel A. The distribution of WGTP log2 ratios at 5 genotypable CNVs. Each histogram of log2 ratios in 270 HapMap individuals exhibits three clusters, each corresponding to a genotype of a biallelic CNV, with the two alleles depicted by broken and complete bars, representing lower and higher copy number alleles. Red lines above each histogram denote log2 ratios in the 12 individuals represented in panel B.
Panel B. Mendelian inheritance of five CNVs in four parent-offspring trios. The individual CNVs were genotyped from WGTP clones: green - Chr8tp-17E9; yellow - Chr1tp-31C8; blue - Chr5tp-22E4; red - Chr6tp-5C12; black - Chr6tp-11A11.
A genome-wide map of copy number variation
The average number of CNVs detected per experiment was 70 and 24 for the WGTP and 500K EA platforms, respectively (Supplementary Tables 8-10). Due to the nature of the comparative analysis, each WGTP experiment detects CNVs in both test and reference genomes, whereas each 500K EA experiment detects CNV in a single genome. The median size of CNVs from the two platforms was 228 kb and 81 kb respectively, and the mean size was 341 kb and 206 kb. Consequently, the average length of the genome shown to be copy number variable in a single experiment is 24 Mb and 5 Mb on the WGTP and 500K EA platforms, respectively. The larger median size of the WGTP CNVs partially reflects inevitable overestimation of CNV boundaries on a platform comprising large-insert clones, as CNV encompassing only a fraction of a clone can be detected, but will be reported as if the whole clone was involved.
By merging overlapping CNVs identified in each individual, we delineated a minimal set of discrete copy number variable regions (CNVRs) among the 270 samples (Figure 3, Supplementary Table 11). We identified 913 CNVRs on the WGTP and 980 CNVRs on the 500K EA platform and mapped their genomic distribution (Figure 4). Approximately half of these CNVRs were called in more than one individual and 43% of all CNVs identified on one platform were replicated on the other. Combining the data resulted in total of 1,447 discrete CNVRs, covering 12% (~360Mb) of the human genome. Using locus-specific quantitative assays on a subset of regions we validated 173 (12%) of these CNVRs (Supplementary Tables 4 & 12). A minority (30%) of these 1,447 CNVRs overlapped those identified in previous studies 1-3,5-8,29. Combining different classes of experimental replication revealed that 957 (66%) of the 1,447 CNVRs detected here have either been replicated on both WGTP and 500K EA platforms, or with a locus-specific assay, or in another individual, or in a previous study (Supplementary Table 12). Whole genome views of CNV show that while common, large-scale CNV is distributed in a heterogeneous manner throughout the genome (Supplementary Figure 6), no large stretches of the genome are exempt from CNV (Figure 4) and the proportion of any given chromosome susceptible to CNV varies from 6% to 19% (Supplementary Figure 7).
Figure 3. Defining copy number variable regions (CNVRs), copy number variants (CNVs) and CNV ends.

Overlapping CNVs called in five individuals are shown schematically for four loci (in blue), dashed lines indicate overlap. Copy number variable regions (CNVRs) represent the union of overlapping CNVs (in green). Independent juxtaposed copy number variants (in black) are identified by requiring that only individual-specific CNVs that overlap by more than a threshold proportion be merged. Intervals encompassing CNV breakpoints (in red) are defined using platform-dependent criteria (Supplementary Methods), and contain a significant paucity of recombination hotspots 75,76 (Supplementary Table 13), which results from the enrichment of segmental duplications within which fewer inferred recombination hotspots reside.
Figure 4. Genomic distribution of copy number variable regions.

The chromosomal locations of 1,447 CNVRs are indicated by lines to either side of ideograms. Green lines denote CNVRs associated with segmental duplications. The length of right-hand lines represents the size of each CNVR. The length of left-hand lines indicates the frequency that a CNVR is detected (minor call frequency among 270 HapMap samples). When both platforms identify a CNVR, the maximum call frequency of the two is shown. For clarity, the dynamic range of length and frequency are log transformed (see scale bars). All data can be viewed at the Database of Genomic Variants (http://projects.tcag.ca/variation/).
Gaps within the reference human genome assembly have an extremely high likelihood of being associated with CNVs; out of the 345 gaps in the build 35 assembly, 48% (164/345) are flanked or overlapped by CNVRs. This finding highlights the complexity in generating a reference sequence in regions of structural dynamism and emphasizes the need for ongoing characterization of these genomic regions.
Comparing the CNVRs identified on the two platforms reveals that the WGTP and 500K EA platforms largely complement one another. The 500K EA platform is better at detecting smaller CNVs (Supplementary Figure 8), whereas the WGTP platform has more power to detect CNVs in duplicated genomic regions (Supplementary Table 13) where 500K EA coverage is poorer 30.
Some CNVRs encompass two or more independent juxtaposed CNVs. For example, a small deletion found in one individual overlapping a much larger duplication in another individual was merged into a single CNVR, despite these representing distinct events. To delineate independent CNVs (CNV ‘events’) we applied more stringent merging criteria to separate juxtaposed CNVs (Figure 3), and identified 1,116 and 1,203 CNVs on the WGTP and 500K EA platforms respectively (Figure 5 and Supplementary Table 11). We classified these CNVs into five types: (i) deletions, (ii) duplications, (ii) deletions and duplications at the same locus, (iv) multi-allelic loci and (v) complex loci whose precise nature was difficult to discern. Due to the inherently relative nature of these comparative data, it was impossible to determine unambiguously the ancestral state for most CNVs, and hence whether they are deletions or duplications. Here we adopted the convention of assuming that the minor allele is the derived allele 31, thus deletions have a minor allele of lower copy number and duplications have a minor allele of higher copy number. Approximately equal numbers of deletions and duplications were identified on the WGTP platform, whereas deletions outnumbered duplications by approximately 2:1 on the 500K EA platform. In addition, 33 homozygous deletions (relative to the reference sequence) identified on the 500K EA platform were experimentally validated with locus-specific assays (Supplementary Table 14). Most (27/33) of these have not been observed in a previous genome-wide survey of deletions 7.
Figure 5. Classes of copy number variants.

CNVs identified from WGTP and 500K EA platforms can be classified from the population distribution of log2 ratios (exemplified with WGTP data) into five different types (see text). Biallelic CNVs (deletions and duplications) can be genotyped if the clusters representing different genotypes are sufficiently distinct. The numbers of each class of CNV identified on WGTP and 500K EA platforms are given, along with the proportion of those CNVs that overlap segmental duplications. The overall proportion of CNVRs overlapping segmental duplications was 20% and 34% on the 500K EA and WGTP platforms, respectively.
To investigate mechanisms of CNV formation, we studied the sequence context of sites of CNV. Non-allelic homologous recombination (NAHR) can generate rearrangements as a result of recombination between highly-similar duplicated sequences 32,33. Segmental duplications are defined as sequences in the reference genome assembly sharing >90% sequence similarity over >1 kb with another genomic location 34,35. We found that 24% of the 1,447 CNVRs were associated with segmental duplications, a significant enrichment (p<0.05). This association results from two factors: (i) rearrangements generated by NAHR and (ii) not all annotated segmental duplications are fixed in humans, but are, in fact, CNVs. This latter point highlights the essentially arbitrary nature of defining segmental duplications on the basis of a single genome sequence (albeit derived from several individuals).
The likelihood of a CNV being associated with segmental duplications depended on its length and its classification: multi-allelic CNVs, complex CNVs, and loci at which both deletions and duplications occurred were strikingly enriched for segmental duplications (Figure 5, Supplementary Figure 9). This is not surprising given the role that NAHR has been shown to play in generating complex structural variation 36, arrays of tandem duplications that vary in size 37 and reciprocal deletions and duplications 38.
The likelihood of a segmental duplication being associated with a CNV was greater for intra-chromosomal duplications than for inter-chromosomal duplications, and was highly correlated with increasing sequence similarity to its duplicated copy (Supplementary Figure 10). NAHR is known to operate mainly on intra-chromosomal segmental duplications and to require 97-100% sequence similarity between duplicated copies 33,39.
This role for NAHR in generating CNVs in duplicated regions of the genome is supported by the enrichment of segmental duplications within intervals that likely contain the breakpoints of the CNV (Figure 3). We identified 88 CNVs from the 500K EA platform and 53 CNVs from the WGTP platform that contain a pair of segmental duplications, one at either end. These pairs of segmental duplications were biased towards high (>97%) sequence similarity, and were more frequently associated with the longest CNVs (Supplementary Figure 11). In addition to segmental duplications, there are other types of sequence homologies that can promote NAHR, for example, dispersed repetitive elements, such as Alu elements 40. We performed an exhaustive search for sequence homology of all kinds 41 and identified 121 CNVs from the 500K EA platform and 223 on the WGTP platform that contain lengths of perfect sequence identity longer than 100bp between either end of the CNV.
Genomic impact of CNV
Deletions are known to be biased away from genes 5, as a result of selection. In contrast, the selective pressures on duplications are poorly understood; the existence of gene families pays testament to positive selection acting on some gene duplications over longer-term evolution 42. We identified the different classes of functional sequence that fell within CNVRs, and tested whether they were significantly enriched or impoverished within these CNVRs (Table 1, Supplementary Table 13, Supplementary Methods).
Table 1. Functional sequences within copy number variable regions (CNVRs).
Statistical significance of the enrichment or paucity of functional sequences within CNVRs was assessed by randomly permuting the genomic location of autosomal CNVRs (Supplementary Methods). Significant observations are shown in bold. Note that both conserved non-coding elements (CNCs) 77 and CNVRs are biased away from genes so an enrichment of CNCs in CNVRs is not unexpected.
| Functional sequence | WGTP CNVRs | 500K EA CNVRs | Merged CNVRs |
|---|---|---|---|
| RefSeq genes | 2,561 | 1,140 ** | 2,909 ** |
| OMIM genes | 251 | 112 ** | 285 |
| Ultra-Conserved Elements | 48 ** | 16 ** | 50 ** |
| Conserved Non-Coding elements | 111,295 * | 81,517 * | 130,352 * |
| non-coding RNAs | 57 | 29 ** | 67 |
significant (p < 0.05) enrichment
significant (p < 0.05) paucity
It is not possible to define precisely the breakpoints of CNVRs; therefore, some of these functional sequences might flank rather than be encompassed by CNVRs. We observed a significant paucity of all functional sequences (with the exception of conserved non-coding sequences 43) in CNVRs detected on the 500K EA platform, which provided highest resolution breakpoint mapping (Table 1). Thus CNVs are preferentially located outside of genes, and ultra-conserved elements (UCEs) in the human genome 44. We attempted to validate experimentally 11 CNVs containing 12 UCEs. While all but two of the CNVs validated, only 2 UCEs actually fell within these CNVs (Supplementary Table 13B), so the selection against CNV at UCEs is likely to be even stronger than this analysis would suggest. Nevertheless, thousands of putatively functional sequences, including known disease-related genes, flank or fall within these CNVs: over half (58%) of the 1,447 CNVRs overlap known RefSeq genes, and more than 99% overlap conserved non-coding sequences 43.
We examined whether deletions or duplications are equally likely to encompass these different classes of functional sequences. We observed that a significantly lower proportion of deletions than duplications (identified on the 500K EA platform) overlap with the OMIM database of disease-related genes (p=0.017, chi-squared) and RefSeq genes (p=1.7×10−9). Thus deletions are biased away from genes with respect to duplications. The same trend was observed with UCEs but their number is too small to provide statistical significance.
If deletions are under stronger purifying selection (which removes deleterious variants from the population) than duplications 8,45, then deletions should, on average, be both less frequent and smaller than duplications. While, on average, deletions were almost three-fold shorter than duplications (43 kb vs. 120 kb from 500K EA), we detected no significant difference in the frequencies with which deletions and duplications were called (p>0.05 using G-test for independence 46 on WGTP data). We note that our length analysis could be confounded if long duplications arise more frequently than long deletions, while our frequency analysis could be confounded if the power to detect duplications was lower as a result of the smaller relative change in copy number (3:2 vs. 2:1).
We identified functional categories of genes that were enriched within CNVs using the Gene Ontology (GO) database (Supplementary Table 15). The most enriched GO category among genes overlapped by the 1,447 CNVRs was cell adhesion. Other highly enriched categories include sensory perception of smell and of chemical stimulus. Interestingly, neurophysiological processes were also a highly enriched GO category. The most highly enriched GO categories within CNVRs overlapped appreciably with those identified in a previous analysis of genes in CNVs 14. Genes found in segmental duplications are known to be biased in terms of GO categories 34, however, an enrichment of cell adhesion genes was also observed within CNVRs not associated with segmental duplications. We also investigated functional categories that are under-represented within CNVs, as these might reveal classes of genes that are more likely to be dosage sensitive. We noted an impoverishment of GO categories relating to cell signaling, cell proliferation and numerous kinase- and phosphorylation-related categories. The impoverishment of these gene functions within CNVs most likely reflects purifying selection acting both against the altered copy number of cell-signaling molecules vital for development and of dosage-sensitive oncogenes or tumour suppressor genes 47 that could predispose to early-onset tumourigenesis.
Copy number variation of medical relevance
In the absence of phenotypic information for HapMap donors, our data are most relevant for highlighting variable regions of the genome that warrant consideration in disease studies, rather than for immediate application to clinical diagnostics.
We found that 286/1,961 (15.6%) genes in the OMIM morbid map overlapped with CNVs (Supplementary Table 16). We observed numerous examples of possible relevance to both Mendelian and complex diseases. For example, the breakpoint region(s) for 12 of 25 loci involved in genomic disorders (which cause 33 different diseases) such as DiGeorge and Williams-Beuren Syndrome 39 were found to be highly polymorphic (Supplementary Table 17). CNVs were also identified within the regions commonly deleted in DiGeorge, Smith-Magenis, Williams-Beuren, Prader-Willi and Angelman Syndromes, which may be relevant for discerning uncharacterized or atypical cases. We also found CNVs at the Spinal Muscular Atrophy and Nephronophthisis loci, as expected, since these diseases are recessive in nature with relatively high carrier frequencies 33. Finally, 39 CNVs were found to reside within 500 kb of the ends of 36 chromosomal arms, which is relevant when assessing sub-telomeric rearrangements in disease.
We found CNVs already known to be responsible for complex traits, including CCL3L1 and FCGR3B 19,20. Some new observations were also documented. Two CEU samples (mother and offspring) manifested a gain of CNV-95 involving the first six exons of DISC1, which is disrupted in schizophrenia 48. CNV-575, encompassing the LPA apolipoprotein A gene, demonstrated population variability, which may influence susceptibility to atherosclerosis. The CRYBB2-CRYBB3 beta-crystallin genes in CNV-1367 were observed as gains and losses in CEU and YRI samples. However, only gains were detected in Asians, leading us to speculate that variability in crystallin copy number may be linked to population differences of onset of age-related cataracts 49. Following a similar rationale, we highlight CNV-507 for possible involvement in sarcoidosis due to its proximity to the BTLN2 gene 50, and CNV-505 in psoriasis susceptibility, since it covers the 6p21.3 - PSORS1 – susceptibility locus 51.
We also highlight challenges in resolving genotype-phenotype correlations in complex CNV regions and how CNV detection can delineate unstable genomic regions (details in Supplementary Note including Supplementary Figure 12). We identified patients with congenital cardiac defects and lens abnormalities 52 that share a deleted or duplicated ~1Mb region of 1q21.1 containing pertinent candidate genes. Duplication of the same interval was observed in other cases with mental retardation 29. Probands can inherit the disease-associated rearrangements from unaffected parents, which underscores the variable penetrance of some diseases resulting from dosage effects 53,54. We found that this locus is highly duplicated, polymorphically inverted, contains assembly gaps, and is flanked by segmental duplications of variable copy number, all features being increasingly observed in CNV regions of the human genome.
Imprint of CNV on SNP genotypes
Deletions perturb patterns of marker genotypes within pedigrees 54 and these patterns highlight the location of such deletions. SNP genotype patterns characteristic of deletions are enrichment of: null genotypes in homozygous deletions, Mendelian discrepancies in families and Hardy-Weinberg disequilibrium within a population 5,7. Duplications can similarly lead to misinterpretation of marker genotypes 53,55, although their impact on high-density SNP maps is poorly understood.
We characterized the patterns of Phase I HapMap SNP genotypes within deletions and duplications on a chromosome-by-chromosome basis to take account of regional biases, and found that most classes of aberrant SNP genotypes were significantly enriched in both deletions and duplications on most chromosomes (Supplementary Figure 13). We replicated the patterns of SNP genotypes within deletions (described above), and demonstrated that duplications also impact significantly upon SNP genotypes. The spectrum of SNP failures enriched within duplications distinguishes them from deletions (Supplementary Figure 13, Supplementary Table 18). Most notably, SNPs exhibiting Mendelian inconsistencies are more common within deletions than duplications, whereas the opposite is true for SNPs in Hardy-Weinberg disequilibrium and SNPs with missing genotypes.
Cell-line artefacts also impact upon SNP genotypes. For example, the partial deletion of 17p in NA12056 causes a distinct paucity of heterozygous SNP genotypes in that individual (one quarter that of the population average) over several megabases of chromosome 17 (HapMap release 20).
Linkage disequilibrium around CNVs: implications for association studies
Indirect methods to identify causative variants, such as co-segregation of linked markers in families and genetic association with markers in linkage disequilibrium (LD) with the causative variant, are considered to be blind to the nature of the underlying mutation 56. This raises the question of whether SNP-based whole genome association studies have the same power to detect disease-related CNVs as for disease-related SNPs. This question can be addressed by considering the maximal pairwise LD (r2) between a particular variant (CNV or SNP) and any of its neighbouring polymorphic markers. If a neighbouring marker is in high LD (r2 close to 1) with the variant of interest, that variant is ‘tagged’ by the neighbouring marker; the genotype of the variant of interest can be predicted with high probability by knowing the genotype at the ‘tagging’ marker.
Recent studies of LD around CNVs have produced conflicting evidence as to the degree to which CNVs are ‘tagged’ by neighbouring SNPs 6-8. Here we performed a balanced comparison between the LD properties of biallelic CNVs and Phase I HapMap SNPs by considering CNVs irrespective of their genomic location, and by analysing CNV genotypes (Supplementary Table 7 and Supplementary Figure 14) of the same frequency and quality as SNP genotypes 5,7,25 (see Methods for details). We quantified pairwise LD around 65 biallelic CNVs using the same three analysis panels as for the Phase I HapMap (CEU, YRI, JPT+CHB). Comparing the proportion of variants ‘tagged’ by a neighbouring SNP with an arbitrary threshold of r2>0.8 shows that whereas 75-80% of Phase I SNPs in non-African populations were tagged, only 51% of CNVs were tagged in the same populations (Figure 6A and Supplementary Table 19). In the YRI, both SNPs and CNVs exhibited lower LD, with only 22% of CNVs being tagged with r2>0.8.
Figure 6. Patterns of linkage disequilibrium between CNVs and SNPs.

Panel A. The proportion of variants that are tagged by a nearby proxy SNP (from Phase I HapMap) increases as the pairwise LD (r2) required for a proxy SNP is relaxed. This cumulative distribution is shown for both Phase I HapMap SNPs and for 65 biallelic CNVs.
Panel B. Histograms of the log2 ratios among all HapMap individuals are shown for thirteen multi-allelic CNVs. The maximal squared Pearson correlation coefficient (R2) observed at a neighbouring Phase I HapMap SNP - which is highly correlated with pairwise LD (r2) at biallelic CNVs (Supplementary Figure 15) - is given for each CNV.
We considered three explanations for these observations of lower apparent LD around CNVs than SNPs. First, some duplications might represent transposition events that would generate LD around the (unknown) acceptor locus but not the donor locus. One of the genotyped CNVs is known to be a duplicative transposition 57, but evidence from de novo pathogenic duplications strongly suggests a preference for tandem, rather than dispersed, duplications, regardless of whether duplication is caused by NAHR 58. Second, some CNVs might undergo recurrent mutations or reversions, especially tandem duplications which are mechanistically prone to unequal crossing over, causing reversions back to a single copy 12. However, duplications were not in lower LD with flanking SNPs than were deletions. Finally, we considered that CNVs might occur preferentially in genomic regions with lower densities of SNP genotypes in HapMap Phase I. We found that CNVs are enriched within segmentally-duplicated regions of the genome, in which there is a paucity of genotyped SNPs due to technical difficulties 25. Thus the strongest factor decreasing apparent LD around biallelic CNVs is not that LD around such CNVs is necessarily lower, but that there is, on average, lower coverage of these structurally dynamic regions of the genome by SNPs genotyped in Phase I of the HapMap project.
We investigated whether the copy number of multi-allelic CNVs could be predicted reliably by nearby SNPs. We treated diploid genome copy number at multi-allelic CNVs as a quantitative trait, and asked which nearby SNPs are most predictive of this trait, and how strongly predictive these SNPs are. We identified 13 multi-allelic CNVs in which the quantitative WGTP data clearly clustered into discrete diploid genome copy numbers and quantified the predictive ability of neighbouring SNPs using the square of Pearson's correlation coefficient (R2) (Supplementary Figure 15). We found that diploid copy number of multi-allelic CNVs is poorly predicted by neighbouring SNPs (Figure 6B). It may be that combining information from several SNPs could provide greater power for predicting diploid genome copy number at these loci.
Population genetics of copy number variation
In contrast to other classes of human genetic variation, the population genetics of copy number variation remains unexplored. The distribution of copy number variation within and among different populations is shaped by mutation, selection and demographic history. A range of polymorphisms, including SNPs 25, microsatellites 59 and Alu insertion variants 60, have been used to investigate population structure. To demonstrate the utility of copy number variation genotypes for population genetic inference we performed population clustering 61 on 67 genotyped biallelic CNVs. We obtained the optimal clustering with the assumption of three ancestral populations, with the African, European and Asian populations clearly differentiated (Figure 7). Population differentiation of individual variants is commonly estimated by the statistic FST, which varies from 0 (undifferentiated) to 1 (population-specific) 62. The average FST for the same 67 autosomal CNVs was 0.11, very similar to that observed for all autosomal Phase I HapMap SNPs (0.13) 25.
Figure 7. Population clustering from CNV genotypes.

A triangle plot showing the clustering of 210 unrelated HapMap individuals assuming three ancestral populations (k=3). The proximity of an individual to each apex of the triangle indicates the proportion of that genome that is estimated to have ancestry in each of the three inferred ancestral populations. The clustering together of most individuals from the same population near a common apex indicates the clear discrimination between populations obtained through this analysis. The clustering was qualitatively similar to that obtained previously with a similar number of biallelic Alu insertion polymorphisms on different African, European and Asian population samples 60.
Recent population-specific positive selection elevates population differentiation. To explore population differentiation at all CNVs, we devised a statistic, VST, that estimates population differentiation based on the quantitative intensity data and varies from 0 to 1, similar to FST (Supplementary Figure 16). Estimating VST for all clones on the WGTP array and all CNVs on the 500K EA array revealed a number of outliers with levels of population differentiation suggestive of population-specific selective pressures (Figure 8 and Supplementary Table 20). Among these outliers were two CNVs previously demonstrated to have elevated population differentiation 7,19: UGT2B17 is a gene encoding a UDP-glucuronosyl transferase with roles in androgen metabolism and xenobiotic conjugation 63,64, and CCL3L1 is a chemokine-encoding multi-copy gene at which greater copy numbers protect against HIV-1 infection 19.
Figure 8. Population differentiation for copy number variation.

Population differentiation, estimated by VST, for each of the three population pairwise comparisons is plotted along each chromosome. For each pairwise comparison, the VST values for all clones on the WGTP platform are shown in the lighter colour with filled circles, with VST values of CNVs detected on the 500K EA platform superimposed in a darker shade with unfilled circles. Histograms showing the distributions of log2 ratios (on the WGTP platform) among the unrelated individuals in each population are plotted for 4 example CNVs exhibiting high population differentiation, labelled A-D. Each example histogram is labelled with the chromosome coordinates of the WGTP clone, and flanking/encompassed genes are given for those CNVs mentioned in the text.
Not all regions that have been under recent positive selection exhibit elevated population differentiation 65. To detect other CNVs that may have recently been under positive selection, we identified CNVRs that fell within 124/752 (16%) of genomic locations previously shown 66 to exhibit haplotype patterns suggestive of a partial selective sweep (Supplementary Table 21). Two of these overlapping CNVs also fell within the set exhibiting highest population differentiation, shown in Table 3. One of these selection-associated CNVs is a duplication specific to the CEU (Figure 8) and lies near to the MAPT gene, which is associated with a set of neurodegenerative disorders known as ‘tauopathies’ 67. Both the MAPT gene and the duplication lie within a chromosomal region that has recently been shown to have a complex evolutionary history, characterized by a common chromosomal inversion, deep divergence between inverted haplotypes and recent positive selection in European populations 68. We adapted methods used to identify partial selective sweeps at SNPs 66,69 to estimate Relative Extended Haplotype Homozygosity values (REHH) on either flank of the 67 genotypable CNVs (Supplementary Methods). We identified no convincing signals (p<0.01 on both flanks) of positive selection on any CNV in any one population, although there were weaker signals (p<0.05) apparent for some CNVs (Supplementary Figure 17 and Supplementary Table 21).
Discussion
Our map of copy number variation in the human genome demonstrates the ubiquity and complexity of this form of genomic variation. The abundance of functional sequences of all types both within and flanking areas of copy number variation suggests that the contribution of CNVs to phenotypic variation is likely to be appreciable. This prediction is underscored by the impact of copy number variation on variation in gene expression (Barbara Stranger and Emmanouil Dermitzakis, personal communication).
CNV assessment should now become standard in the design of all studies of the genetic basis of phenotypic variation, including disease susceptibility. Similarly important will be CNV annotation in all future genome assemblies. The identification of CNVs causing severe, sporadic diseases has been hampered by an inability to distinguish between normal and causative variants. Our CNV map, in tandem with the DECIPHER (http://www.sanger.ac.uk/PostGenomics/decipher/) project's sharing of copy number information on patients with rare, severe phenotypes should advance progress in this area. For Mendelian genetic diseases, our data contain numerous known and candidate recessive disease alleles, and existing linkage data can be used to prioritise CNVs for further investigation.
Genetic association studies are the predominant strategy for identifying haplotypes conferring risk for complex genetic diseases. Such studies are typically based on SNP genotyping, either within candidate loci or genome-wide 56. Our analysis of linkage disequilibrium between CNVs and SNPs gives us limited optimism that CNVs influencing risk to complex disease will be detected by such approaches. The tag-SNPs that we have identified (Supplementary table 19) for specific CNVs can be used as proxies for these CNVs. Moreover, CNV-specific genotyping assays can be developed for CNVs for which tag-SNPs are not readily identifiable but whose proximity to candidate genes warrants further characterization. Finally, we see great merit in mining CNV information from quantitative SNP genotyping data, and in enriching future generations of genome-wide SNP genotyping platforms by targeting untaggable CNVs.
The overall utility of any map depends on its coverage and its completeness. Extrapolation based on existing data suggests that smaller deletions (<20 kb) are much more frequent than larger deletions (>20 kb) 5, and the same may be true for duplications. While we have generated the most complete CNV map yet described, given our lower power to detect smaller CNVs a substantial fraction of copy number variation >1 kb in size in these individuals remains to be characterized. No single available technology will capture all variation. Smaller rearrangements are amenable to detection using technologies such as sequence assembly comparisons 70, paired-end sequence relationships 4, sequence trace analysis 71 and higher-resolution tiling arrays 5. Ultimately, it is desirable to know the precise chromosomal location and sequence content of each and every CNV. At present, generating this information requires the use of multiple experimental methods. However, in the future, the comparison of independently assembled whole genome sequences could provide a definitive solution.
Our study of human genetic variation ties together cytogenetics, sub-microscopic copy number variation and single nucleotide polymorphisms, providing a framework for future genetic studies. This framework will need to be supported by continual refinement of the reference genome sequence and robust nomenclature and databasing for structural variation - both facilitated through international collaboration - to enable further unraveling of the complexity of human genomic variation.
Methods
CNV determination on WGTP and 500K EA arrays
The experimental methods and algorithms used for CNV calling using the WGTP and the 500K EA platforms are described in the Supplementary Methods and in two accompanying papers 26,28.
500K EA and WGTP data quality assessment
In order to estimate false positive and false negative rates on both platforms, we used quantitative PCR to experimentally test CNVs called from replicate experiments with NA15510 4 (Supplementary Tables 1-4). With 500K EA, the average proportion of CNV calls from this sample that were false positive was 2.3% (0.33 out of 15 CNV calls), and the false negative percentage was 24% (3.3 validated CNVs not called in any one replicate/ 38 total validated CNVs) 28. With WGTP, we found an average false positive proportion of 5% (3.4 out of 68.2) and a false negative percentage of 37.8% (58 non called out of 154 tested) 26. Reproducibility was also assessed by analyzing 10 HapMap DNAs in triplicate (Supplementary Table 6). For the 500K EA platform, on average, 80% of CNVs were called in all three replicates, 10% were called twice, and 10% were called only once. For the WGTP platform, using the replicate with the lowest number of calls as the baseline, on average 73% of CNVs were called in 3 experiments, 14% were called twice, and 13% were called once 26.
Population genetic and statistical analysis
Sixty-seven non-redundant biallelic CNVs suitable for genotyping were identified on the WGTP and 500K EA platforms. Two procedures were used to cluster intensity ratios into discreet copy number genotypes: Kmeans and Partitioning Around Medoids (PAM) 72. Pairwise LD (r2) was estimated between 65 biallelic CNVs and all filtered non-redundant Phase I HapMap SNPs within 500 kb of the CNV borders using HaploView 73. Population clustering was performed using STRUCTURE 61 and population specific CNVs were estimated using FST 62 and the new statistic VST (Supplementary Figure 16). VST is calculated by considering (VT−VS)/VT where VT is the variance in log2 ratios apparent among all unrelated individuals and VS is the average variance within each population, weighted for population size. For REHH analysis 69,74, we treated each genotyped CNV as a SNP located at the CNV end, and used Phase I HapMap SNPs 500 kb upstream and downstream of the CNV using the program Sweep (http://www.broad.mit.edu/mpg/sweep/resources.html). See Supplementary Methods for more details.
Data release
The raw data from the 500K EA as well as the 500K commercial arrays are posted at the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/), with accession numbers GSE5013 and GSE5173. WGTP data is posted at ArrayExpress (www.ebi.ac.uk/arrayexpress/) with accession number E-TABM-107, and at the Wellcome Trust Sanger Institute (www.sanger.ac.uk/humgen/cnv/data/). CNV calls have been released at the Database of Genomic Variants (http://projects.tcag.ca/variation/) integrated with all other CNV data.
Supplementary Material
Acknowledgements
The authors thank Christine Bird, Yuan Chen, Mark Daly, Ciara Fahey, Ann M. Joseph-George, Yongshu He, Kahori Hirose, Zhizhou Hu, Vikram Jayanth, Cordelia Langford, Martin Li, Chao Lu, Guoying Liu, Zhanquin Liu, Hiroko Meguro, Lorena Pantano, Tara Paton, Itsik Pe'er Sanjeev Pullenayegum, Ying Qi, Simone Russell, Mark Schachowsky, Mary Shago, Kaori Shiina and Yali Xue for advice, sharing data, technical assistance or bioinformatics support. The Centre for Applied Genomics at The Hospital for Sick Children and the Microarray Facility of the Wellcome Trust Sanger Institute are acknowledged for database support and array printing, respectively. The authors thank James R. Lupski and Jonathan Pritchard for insightful comments on earlier versions of the manuscript. The research was supported by The Wellcome Trust (MEH, NPC, CTS), Canada Foundation of Innovation and Ontario Innovation Trust (SWS), Canadian Institutes of Health Research (CIHR)(SWS), Genome Canada/Ontario Genomics Institute (SWS), the McLaughlin Centre for Molecular Medicine (SWS), Ontario Ministry of Research and Innovation (SWS), the Hospital for Sick Children Foundation (SWS), The Department of Pathology, Brigham and Women's Hospital (CL), The Leukemia and Lymphoma Society (CL), the Core Research for Evolutional Science and Technology (CREST) from the Japan Science and Technology Agency, Grant-in-Aid for Scientific Research (S)(HA), Grants-in-Aid for Young Scientists (B) and Scientific Research on Priority Areas “Applied Genomics” from the Ministry of Education, Culture, Sports, Science and Technology of Japan (SI), the “Departament d'Universitats Recerca i Societat de la Informació” (SGR2005-00008) (XE) and the Genoma España and Genome Canada joint R+D+I projects (XE and SWS), the National Genotyping Center Barcelona Node (CeGen) supported by Genoma España (XE), and the Instituto de Salud Carlos III (CIBER-CB06/03/0034) (XE), and a Packard Fellowship to J. K. Pritchard (DC). LF is supported by a fellowship from CIHR, RR by a Sanger Institute Postdoctoral Fellowship and AC from the Natural Science and Engineering Research Council. SWS is an Investigator of the CIHR and International Scholar of the Howard Hughes Medical Institute.
References
- 1.Iafrate AJ, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–51. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 2.Sebat J, et al. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–8. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 3.Sharp AJ, et al. Segmental duplications and copy-number variation in the human genome. Am J Hum Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tuzun E, et al. Fine-scale structural variation of the human genome. Nat Genet. 2005;37:727–732. doi: 10.1038/ng1562. [DOI] [PubMed] [Google Scholar]
- 5.Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK. A high-resolution survey of deletion polymorphism in the human genome. Nat Genet. 2006;38:75–81. doi: 10.1038/ng1697. [DOI] [PubMed] [Google Scholar]
- 6.Hinds DA, Kloek AP, Jen M, Chen X, Frazer KA. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat Genet. 2006;38:82–5. doi: 10.1038/ng1695. [DOI] [PubMed] [Google Scholar]
- 7.McCarroll SA, et al. Common deletion polymorphisms in the human genome. Nat Genet. 2006;38:86–92. doi: 10.1038/ng1696. [DOI] [PubMed] [Google Scholar]
- 8.Locke DP, et al. Linkage Disequilibrium and Heritability of CNPs within Duplicated Regions of the Human Genome. Am J Hum Genet. 2006;79:275–290. doi: 10.1086/505653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fredman D, et al. Complex SNP-related sequence variation in segmental genome duplications. Nat Genet. 2004;36:861–6. doi: 10.1038/ng1401. [DOI] [PubMed] [Google Scholar]
- 10.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 11.Freeman JL, et al. Copy Number Variation: New Insights in Genome Diversity. Genome Research. 2006;16:949–961. doi: 10.1101/gr.3677206. [DOI] [PubMed] [Google Scholar]
- 12.Bridges CB. The Bar ‘gene’: a duplication. Science. 1936;83:210–211. doi: 10.1126/science.83.2148.210. [DOI] [PubMed] [Google Scholar]
- 13.Buckland PR. Polymorphically duplicated genes: their relevance to phenotypic variation in humans. Ann Med. 2003;35:308–15. doi: 10.1080/07853890310001276. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen DQ, Webber C, Ponting CP. Bias of Selection on Human Copy-Number Variants. PLoS Genet. 2006;2:e20. doi: 10.1371/journal.pgen.0020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Repping S, et al. High mutation rates have driven extensive structural polymorphism among human Y chromosomes. Nat Genet. 2006;38:463–7. doi: 10.1038/ng1754. [DOI] [PubMed] [Google Scholar]
- 16.Inoue K, Lupski JR. Molecular mechanisms for genomic disorders. Annu Rev Genomics Hum Genet. 2002;3:199–242. doi: 10.1146/annurev.genom.3.032802.120023. [DOI] [PubMed] [Google Scholar]
- 17.Lupski JR, Stankiewicz P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005;1:e49. doi: 10.1371/journal.pgen.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw-Smith C, et al. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J Med Genet. 2004;41:241–8. doi: 10.1136/jmg.2003.017731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gonzalez E, et al. The influence of CCL3L1 gene-containing segmental duplications on HIV-1/AIDS susceptibility. Science. 2005;307:1434–40. doi: 10.1126/science.1101160. [DOI] [PubMed] [Google Scholar]
- 20.Aitman TJ, et al. Copy number polymorphism in Fcgr3 predisposes to glomerulonephritis in rats and humans. Nature. 2006;439:851–5. doi: 10.1038/nature04489. [DOI] [PubMed] [Google Scholar]
- 21.Jongmans MC, et al. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet. 2006;43:306–14. doi: 10.1136/jmg.2005.036061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singleton AB, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 23.Rovelet-Lecrux A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–6. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 24.Feuk L, Marshall CR, Wintle RF, Scherer SW. Structural variants: changing the landscape of chromosomes and design of disease studies. Hum Mol Genet. 2006;15(Suppl 1):R57–66. doi: 10.1093/hmg/ddl057. [DOI] [PubMed] [Google Scholar]
- 25.IHMC A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fiegler H, et al. Accurate and reliable high-throughput detection of copy number variation in the human genome. Genome Res. doi: 10.1101/gr.5630906. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ylstra B, van den Ijssel P, Carvalho B, Brakenhoff RH, Meijer GA. BAC to the future! or oligonucleotides: a perspective for micro array comparative genomic hybridization (array CGH) Nucleic Acids Res. 2006;34:445–50. doi: 10.1093/nar/gkj456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Komura D, et al. Genome-Wide Detection of Human Copy Number Variants using High Density DNA Oligonucleotide Arrays. Genome Res. doi: 10.1101/gr.5629106. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Vries BB, et al. Diagnostic genome profiling in mental retardation. Am J Hum Genet. 2005;77:606–16. doi: 10.1086/491719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wirtenberger M, Hemminki K, Chen B, Burwinkel B. SNP microarray analysis for genome-wide detection of crossover regions. Hum Genet. 2005;117:389–97. doi: 10.1007/s00439-005-1323-y. [DOI] [PubMed] [Google Scholar]
- 31.Marth GT, Czabarka E, Murvai J, Sherry ST. The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics. 2004;166:351–72. doi: 10.1534/genetics.166.1.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurles M. How homologous recombination generates a mutable genome. Hum Genomics. 2005;2:179–86. doi: 10.1186/1479-7364-2-3-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet. 2002;18:74–82. doi: 10.1016/s0168-9525(02)02592-1. [DOI] [PubMed] [Google Scholar]
- 34.Bailey JA, et al. Recent segmental duplications in the human genome. Science. 2002;297:1003–7. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 35.Cheung J, et al. Genome-wide detection of segmental duplications and potential assembly errors in the human genome sequence. Genome Biol. 2003;4:R25. doi: 10.1186/gb-2003-4-4-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aradhya S, et al. Multiple pathogenic and benign genomic rearrangements occur at a 35 kb duplication involving the NEMO and LAGE2 genes. Hum Mol Genet. 2001;10:2557–67. doi: 10.1093/hmg/10.22.2557. [DOI] [PubMed] [Google Scholar]
- 37.Neitz M, Neitz J. Numbers and ratios of visual pigment genes for normal red-green color vision. Science. 1995;267:1013–6. doi: 10.1126/science.7863325. [DOI] [PubMed] [Google Scholar]
- 38.Chance PF, et al. 2 Autosomal-Dominant Neuropathies Result from Reciprocal DNA Duplication/Deletion of a Region on Chromosome-17. Hum Mol Genet. 1994;3:223–228. doi: 10.1093/hmg/3.2.223. [DOI] [PubMed] [Google Scholar]
- 39.Lupski JR, Stankiewicz P, editors. Genomic Disorders: the genomic basis of disease. Humana Press; Totawa, New Jersey: 2006. [DOI] [PubMed] [Google Scholar]
- 40.Deininger PL, Batzer MA. Alu repeats and human disease. Mol Genet Metab. 1999;67:183–93. doi: 10.1006/mgme.1999.2864. [DOI] [PubMed] [Google Scholar]
- 41.Kurtz S, et al. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29:4633–42. doi: 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hurles M. Gene duplication: the genomic trade in spare parts. PLoS Biol. 2004;2:E206. doi: 10.1371/journal.pbio.0020206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drake JA, et al. Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nat Genet. 2006;38:223–7. doi: 10.1038/ng1710. [DOI] [PubMed] [Google Scholar]
- 44.Bejerano G, et al. Ultraconserved elements in the human genome. Science. 2004;304:1321–5. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- 45.Brewer C, Holloway S, Zawalnyski P, Schinzel A, FitzPatrick D. A chromosomal duplication map of malformations: regions of suspected haplo- and triplolethality–and tolerance of segmental aneuploidy–in humans. Am J Hum Genet. 1999;64:1702–8. doi: 10.1086/302410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sokal RR, Rohlf JF. Biometry. W.H. Freeman and Co; 1994. [Google Scholar]
- 47.Futreal PA, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–83. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Millar JK, et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 2005;310:1187–91. doi: 10.1126/science.1112915. [DOI] [PubMed] [Google Scholar]
- 49.Sasaki K, Sasaki H, Jonasson F, Kojima M, Cheng HM. Racial differences of lens transparency properties with aging and prevalence of age-related cataract applying a WHO classification system. Ophthalmic Res. 2004;36:332–40. doi: 10.1159/000081636. [DOI] [PubMed] [Google Scholar]
- 50.Valentonyte R, et al. Sarcoidosis is associated with a truncating splice site mutation in BTNL2. Nat Genet. 2005;37:357–64. doi: 10.1038/ng1519. [DOI] [PubMed] [Google Scholar]
- 51.Orru S, Giuressi E, Carcassi C, Casula M, Contu L. Mapping of the major psoriasis-susceptibility locus (PSORS1) in a 70-Kb interval around the corneodesmosin gene (CDSN) Am J Hum Genet. 2005;76:164–71. doi: 10.1086/426948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Christiansen J, et al. Chromosome 1q21.1 contiguous gene deletion is associated with congenital heart disease. Circ Res. 2004;94:1429–35. doi: 10.1161/01.RES.0000130528.72330.5c. [DOI] [PubMed] [Google Scholar]
- 53.Lupski JR, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66:219–32. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 54.Chance PF, et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72:143–51. doi: 10.1016/0092-8674(93)90058-x. [DOI] [PubMed] [Google Scholar]
- 55.Matise TC, et al. Detection of tandem duplications and implications for linkage analysis. Am J Hum Genet. 1994;54:1110–21. [PMC free article] [PubMed] [Google Scholar]
- 56.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 57.Wong Z, Royle NJ, Jeffreys AJ. A novel human DNA polymorphism resulting from transfer of DNA from chromosome 6 to chromosome 16. Genomics. 1990;7:222–34. doi: 10.1016/0888-7543(90)90544-5. [DOI] [PubMed] [Google Scholar]
- 58.Woodward KJ, et al. Heterogeneous duplications in patients with Pelizaeus-Merzbacher disease suggest a mechanism of coupled homologous and nonhomologous recombination. Am J Hum Genet. 2005;77:966–87. doi: 10.1086/498048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rosenberg NA, et al. Genetic structure of human populations. Science. 2002;298:2381–5. doi: 10.1126/science.1078311. [DOI] [PubMed] [Google Scholar]
- 60.Bamshad MJ, et al. Human population genetic structure and inference of group membership. Am J Hum Genet. 2003;72:578–89. doi: 10.1086/368061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–59. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- 63.Turgeon D, Carrier JS, Chouinard S, Belanger A. Glucuronidation activity of the UGT2B17 enzyme toward xenobiotics. Drug Metab Dispos. 2003;31:670–6. doi: 10.1124/dmd.31.5.670. [DOI] [PubMed] [Google Scholar]
- 64.Wilson W, 3rd, et al. Characterization of a common deletion polymorphism of the UGT2B17 gene linked to UGT2B15. Genomics. 2004;84:707–14. doi: 10.1016/j.ygeno.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 65.Xue Y, et al. Spread of an inactive form of caspase-12 in humans is due to recent positive selection. Am J Hum Genet. 2006;78:659–70. doi: 10.1086/503116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rademakers R, Cruts M, van Broeckhoven C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum Mutat. 2004;24:277–95. doi: 10.1002/humu.20086. [DOI] [PubMed] [Google Scholar]
- 68.Stefansson H, et al. A common inversion under selection in Europeans. Nat Genet. 2005;37:129–37. doi: 10.1038/ng1508. [DOI] [PubMed] [Google Scholar]
- 69.Sabeti PC, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–7. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- 70.Khaja R, et al. Genome assembly comparison to identify structural variants in the human genome. Nat Genet. doi: 10.1038/ng1921. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bennett EA, Coleman LE, Tsui C, Pittard WS, Devine SE. Natural genetic variation caused by transposable elements in humans. Genetics. 2004;168:933–51. doi: 10.1534/genetics.104.031757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kaufman L, Rousseeuw PJ. Finding groups in data: an introduction to cluster analysis. Wiley; New York: 1990. [Google Scholar]
- 73.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 74.Walsh EC, et al. Searching for signals of evolutionary selection in 168 genes related to immune function. Hum Genet. 2006;119:92–102. doi: 10.1007/s00439-005-0090-0. [DOI] [PubMed] [Google Scholar]
- 75.Petes TD. Meiotic recombination hot spots and cold spots. Nat Rev Genet. 2001;2:360–9. doi: 10.1038/35072078. [DOI] [PubMed] [Google Scholar]
- 76.Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. A fine-scale map of recombination rates and hotspots across the human genome. Science. 2005;310:321–4. doi: 10.1126/science.1117196. [DOI] [PubMed] [Google Scholar]
- 77.Dermitzakis ET, et al. Comparison of human chromosome 21 conserved nongenic sequences (CNGs) with the mouse and dog genomes shows that their selective constraint is independent of their genic environment. Genome Res. 2004;14:852–9. doi: 10.1101/gr.1934904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.