

Abstract
The present work offers an analysis of the historical development of the discovery and use of barbiturates in the field of psychiatry and neurology, a century after their clinical introduction. Beginning with the synthesis of malonylurea by von Baeyer in 1864, and up to the decline of barbiturate therapy in the 1960s, it describes the discovery of the sedative properties of barbital, by von Mering and Fischer (1903), the subsequent synthesis of phenobarbital by this same group (1911), and the gradual clinical incorporation of different barbiturates (butobarbital, amobarbital, secobarbital, pentobarbital, thiopental, etc). We describe the role played in therapy by barbiturates throughout their history: their traditional use as sedative and hypnotic agents, their use with schizophrenic patients in so-called “sleep cures” (Klaesi, Cloetta), the discovery of the antiepileptic properties of phenobarbital (Hauptmann) and their use in the treatment of epilepsy, and the introduction of thiobarbiturates in intravenous anesthesia (Lundy, Waters). We also analyze, from the historical perspective, the problems of safety (phenomena of dependence and death by overdose) which, accompanied by the introduction of a range of psychoactive drugs in the 1950s, brought an end to barbiturate use, except in specific applications, such as the induction of anesthesia and the treatment of certain types of epileptic crisis.
Keywords: barbiturates, history of medicine, sedative-hypnotic drugs, “sleep cures”, epilepsy, anesthesia
Introduction
Throughout the history of humanity, numerous therapeutic agents have been employed for their hypnotic and/or sedative properties, though the true effectiveness of many of them has been fairly limited (Alamo et al 1998). It suffices to mention alcohol itself (in different forms, such as hydromel or wine) or the alkaloids of opium and other narcotic plants (hemp, jimsonweed, belladonna, henbane, etc). More recently, around the late 19th and early 20th centuries, agents such as paraldehyde, chloral hydrate, and bromides were used, until the discovery, at the beginning of the 20th century, of the sedative and hypnotic properties of barbiturates, thanks to the prior synthesis of malonylurea by Adolf von Baeyer in 1864.
The clinical introduction of barbiturates begun a century ago (1904) when the Farbwerke Fr Bayer and Co brought onto the market the first agent of this type, diethyl-barbituric acid, giving rise to profound changes in the pharmacological approach to the psychiatric and neurological disorders of the time. A large number of previously untreatable patients gained access to treatment and improved their prognosis. The most significant results were obtained in the treatment of patients with serious neuroses and psychoses and with severe emotional repression, who as a result of being administered barbiturates, especially intravenously, overcame their inhibitions, thus facilitating psychotherapeutic treatment. Barbiturates were also useful in the treatment of sleep disorders as well as being the first truly effective pharmacological tools for the management of epileptic seizures. Furthermore, they opened up the field of intravenous anesthesia, playing a prominent role in anesthetic induction, above all for minor operations.
In the course of the 20th century, more than 2500 barbiturates were synthesized, 50 of which were eventually employed clinically. Their use was widespread and many still have some use today. One hundred years after the introduction in clinical pharmacology of the original compound, oxybarbiturates, in general, continue to be the selected drugs in the treatment of some serious forms of insomnia and in some types of epilepsy. Similarly, some thiobarbiturates and some ultrashort-acting barbiturates are still used today as inducers of general anesthesia. Nevertheless, currently, 5 or 6 derivates of barbiturates are sufficient to cover the therapeutic applications that still require them.
Sedative and anticonvulsant drugs in the pre-barbiturate era
Although, as mentioned, the therapeutic agents historically employed for their sedative, hypnotic, or anticonvulsant effects have been quite numerous, the most specific drugs in this regard have their origin in the 19th century. Such is the case of choral hydrate, different alkaloids and, above all, bromides (Hollister 1983; Sneader 1985; Scott 1992; Lehmann 1993; Shorvon and Sander 1996; Shorter 1997; Alamo et al 1998; Healy 2002).
The second half of the 19th century is called by some authors, such as Shorter (1997), the “alkaloids era”. Alkaloids were introduced into psychiatry as sedatives and hypnotics, thanks to the isolation of morphine from opium, in 1805, by the German pharmacist Friedrich Sertürner. In 1861, Wilhelm Griesinger, in the second edition of his Die Pathologie und Therapie der Psychischen Krankheiten, defended the use of opium in sleep disorders, pointing out the improvements it brought about in patients suffering from anxiety. However, the alkaloids that met with most success were those isolated from different species of the Solanaceae family: plants known for their hallucinogenic effects, such as hyoscyamus, whose sedative and hypnotic properties were described by the Viennese pharmacologist Karl Schroff in 1868. In 1839, chemists at the E Merck company in Darmstadt (Germany) had already isolated hyoscyamine, another alkaloid, which became popular in the late 19th century, forming part of many of the “cocktails” administered in neuropsychiatric institutions at that time (Woodward 1994). Finally, the year 1880 saw the isolation of hyoscine (called scopolamine in North America), an alkaloid that was also widely used in psychiatric cocktails, such as the famous Hyoscine Co A, which contained hyoscine, morphine, and atropine, and was administered to highly excited and aggressive manic patients (Norton 1979).
The first drug that could truly be called hypnotic is chloral hydrate. Synthesized in 1832 by Justus von Liebig, a chemist from Giessen, it was not analyzed as a hypnotic until 1869 by the Berlin pharmacologist Oskar Liebreich. The hypothetical mechanism to which its action was ascribed was based on the mistaken belief that, in vivo, chloral hydrate was capable of transforming itself into formic acid and chloroform, whose properties were already known at that time (Sourkes 1992). Very soon, chloral hydrate substituted morphine and the Solanaceae alkaloids, given its convenience, as it could be administered without the need for injection, allowing treatment in the home and making it unnecessary to confine patients to neuropsychiatric institutions (Shorter 1997).
Nevertheless, it would be the bromides that were most widely used in the second half of the 19th century, either as sedatives or for the treatment of epilepsy, having been introduced for these applications by the internist and obstetrician Sir Charles Locock in 1857. It was in that year that Locock reported his results in the treatment with bromides in women with what the author has named as catamenial or hysteriform epileptic seizures, obtaining positive outcomes in 14 women out of a sample of 15. From that time on, bromides were widely introduced in asylums and similar institutions throughout Europe, given their sedative and antiepileptic properties, the relevant function in the latter case being to reduce the expression of the epileptic patients’ sexuality. Another contribution in relation to the neuropsychiatric use of bromides was made by the British doctor Neil MacLeod, who in 1897, while working in Shanghai, carried out the first “sleep cure” with these salts. MacLeod called it “the bromide sleep” (MacLeod 1900), and some authors, such as Shorter (1997), have considered this technique as the first pharmacological therapy that, within psychiatry, succeeded in improving the symptoms of psychiatric patients. However, the main problem with bromides resided in their high toxicity (neurological and gastrointestinal disorders, irritability, hallucinations, deliria, and lethargy), given their long half-life (elimination taking around 12 days) and their capacity for accumulation in tissue; as a result, they were gradually phased out after the introduction of barbiturates in the early part of the 20th century (Balme 1976).
Other substances used as hypnotics and sedatives and eventually as anticonvulsants were also introduced in the 19th century and the early decades of the 20th century. Such is the case of paraldehyde, discovered by Wildenbusch in 1829 and introduced into clinical practice by Vincenzo Cervello in 1882; and sulphonal, whose hypnotic action was discovered by chance by Eugen Baumann and Alfred Kast in 1887 (Kast 1888). Finally, those seeking to treat epilepsy turned, as well as to potassium bromide, chloral hydrate, or hyoscine, to a whole host of substances of more questionable efficacy, including opium, belladonna, atropine, stramonium, strophanthus, cannabis indica, and zinc oxide.
The discovery and clinical introduction of barbiturates as sedative and hypnotic agents
Between the 1920s and the mid-1950s, practically the only drugs used as sedatives and hypnotics were barbiturates (Lehmann and Ban 1970). From a chemical point of view, these drugs are closed-chain ureic compounds, whose nucleus is malonylurea (a combination of urea, a product present in animal excrement, and malonic acid, an acid derivative taken from apples) (Figure 1). Barbiturates were synthesized in 1864 by Adolf von Baeyer, though the synthetic process was developed and perfected by the French chemist Edouard Grimaux in 1879, making possible the subsequent widespread development of barbiturate derivatives (Carter 1951). Von Baeyer, a disciple of Robert W Bunsen and Friedrich A Kekulé, taught at the universities of Strasbourg and Munich, was the founder of what was to become the Bayer Chemical Co, and received the Nobel Prize in Chemistry in 1905 for his contribution to the development of organic chemistry (Figure 2a).
Figure 1.
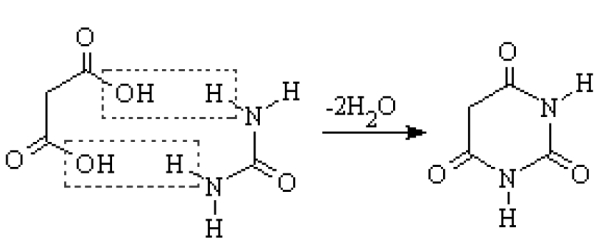
Synthesis of barbituric acid, from the combination of malonic acid (left) and urea (right).
Figure 2.

(a) Adolf von Baeyer (1835–1917); (b) Josef von Mering (1849–1908); (c) Emil Fischer (1852–1919).
There are various hypotheses about the origin of the term “barbiturates” (Dundee and McIlroy 1982). According to one of these, Baeyer may have used this name for the compounds for sentimental reasons, in honor of his friend Barbara (Cohen 1943). Other authors, however, claim that the name derives from the fact that Baeyer celebrated his discovery in a tavern near his home that was frequented by artillery officers, who themselves were celebrating the day of their patron, St Barbara (Sharpless 1970). A third possibility is that the term is inspired by the “barbed” appearance of the crystals of these ureic compounds (Fieser 1944). In any case, it is clear that the union of the elements “barb(ara)” and “urea” forms the basis of the name.
From malonylurea to barbital
The first of the barbiturates to come onto the market was diethyl-barbituric acid, also known as barbital, malonal, or gardenal. Synthesized in 1881 by Conrad and Guthzeit, on treating the argentic salt of barbituric acid with ethyl iodide, it was introduced clinically as a hypnotic by the German companies E Merck (Darmstadt) and F Bayer and Co (Elberfeld) in 1904, thanks to the work of Josef Freiherr von Mering (Figure 2b) and Emil Fischer (Nobel Prize in Chemistry, 1902) (Figure 2c).
Von Mering, who taught pharmacology at the University of Halle, had observed that some of the synthetic compounds obtained towards the end of the 19th century and commercialized as hypnotics, such as sulphonal, contained in their molecular structure a carbon atom with two ethyl groups. Furthermore, knowing of von Baeyer’s work with derivatives of urea, von Mering decided to study the hypnotic properties of diethyl-acetylurea, and found that it was even more potent than sulphonal. The next step was to analyze the properties of 5,5-diethyl-barbituric acid, for which he turned to Fischer, an old friend from his student days. At that time, Fischer, doyen of the German organic chemists, was Professor of Chemistry at the University of Berlin. Moreover, Fischer was well acquainted with the chemistry of malonylurea, as he had been von Baeyer’s assistant in Munich for eight years. Together with his nephew Alfred Dilthey, he tested the new, resynthesized product, demonstrating, in dogs, that its hypnotic power was far greater than that of von Mering’s diethyl-acetylurea (Sneader 1985). When Fischer told his friend von Mering about this finding, the latter happened to be in the Italian city of Verona, and it was this that prompted him to call the new drug Veronal® (Cohen 1943; Sharpless 1970). Nevertheless, other authors argue that the name Veronal (from Latin, verus = true) was coined by Fischer, who claimed to have found the “true” hypnotic compound (Sneader 1985). This new hypnotic drug was patented by Fischer in January 1903, and two months later the first scientific data on barbiturates were published in a brief report (Fischer and von Mering 1903). The licence for its commercialization in the USA was granted to the Winthrop Chemical Company.
The term barbital for diethyl-barbituric acid is a later development, coming as a result of the economic effects of World War I. After the United States entered the conflict, in 1917, Congress passed the Trading with the Enemy Act 1917, which permitted them as a kind of war booty to manufacture German products protected by patent, modifying their generic name and with the profits going to the American subsidiaries of the German companies (Sneader 1985). Thus, the American Medical Association approved the name barbital, whilst in the United Kingdom, through a similar mechanism, diethyl-barbituric acid came to be called barbitone. From this point on the two endings “-al” and “-one” could be found in the nomenclature of barbiturates.
Veronal had hypnotic, sedative, and anticonvulsant properties (Figure 3a). It could calm manic patients and help melancholic patients to sleep, and was an effective inducer of sleep in insomniacs. The first trials with barbital were carried out by Hermann von Husen (1904), a young psychiatrist affected by sleep disorders, who tried the new drug on himself. After taking 0.5 g of Veronal the first night and 1 g the following night, he reports:
Figure 3.

(a) Elixir Veronal from Dr Bustamante’s Laboratories it is a “Practical treatment of insomnia”. They have also added audaciously “Secure and harmless”. Finally they say that “it tastes good and acts smoothly, being absorbed by the organism”. (b) Advertisement for Abbott sodium pentobarbital in an American medical journal of 1933, highlighting its “short but powerful hypnotic effect and prolonged sedative action from small dosage”.
In both cases, after 10–15 minutes, I fell into a growing state of dejection that led to deep sleep after around 30 minutes. After half a gram of Veronal I slept for 8 hours, and after a whole gram, around 9 hours. On the first morning I awoke fresh and rested; on the second morning, after the higher dose, I found it difficult to get out of bed (von Husen 1904, p 59).
The consolidation of barbiturate therapy: phenobarbital
By means of small modifications to the chemical structure of the barbituric acid molecule, more than 2500 different agents were synthesized. The first barbital analogs, numbering around 18, were synthesized and tested by the group made up of von Mering, Fischer, and Dilthey. One of them, perhaps that most widely used subsequently, was phenobarbital, synthesized by Hörlein in 1911, on substituting one of the ethyl groups by a phenyl radical. Phenobarbital was employed in therapy as a hypnotic for the first time in 1912 by Loewe, Juliusburger, and Impens, and that same year it was commercialized by F Bayer and Co, under the name Luminal®. Phenobarbital, with a more prolonged pharmacological action than its predecessor, soon became “king of the barbiturates”, both in hospitals and in outpatient care (Shorter 1997). This drug opened up the way, moreover, to another important therapeutic application of barbiturates, as will be mentioned later: the treatment of epilepsy.
Both Veronal (barbital) and Luminal (phenobarbital), the first two representatives of the series of barbiturates, were accepted by the international pharmacopoeia, such as the United States Pharmacopoeia (USP X) in 1926, and the British Pharmacopoeia in 1914 and 1932, respectively. Later, both drugs were also included in the Pharmacopoeia Internationalis.
Clinical introduction of the new barbiturates
The new barbiturates brought substantial advantages compared with their classical predecessors, such as a greater potency and duration of action, as well as a wider therapeutic range. However, of the several thousand that were synthesized, only about 50 came onto the market, and of these no more than a couple of dozen were regularly used in clinical practice. The next barbiturate to be used successfully in therapy was butobarbital, whose history begins in World War I. The British war effort required large quantities of acetone for the manufacture of explosives (Sneader 1985), and one of the solutions was provided by Chaim Weizmann, who would later become the first president of the state of Israel. Weizmann found that the bacteria Clostridium acetobutylicum was capable of transforming materials rich in starch into acetone and butyric alcohol, and at low industrial cost. After the war, the cost of butyric alcohol, a chemical that was as useful as it was expensive, fell drastically, thus permitting its use for obtaining numerous synthetic drugs. In 1920, Roger Adams (Abbott Laboratories, Chicago, USA) synthesized the ester of 5-butyl-5-ethyl-malonic acid, an intermediate stage in the synthesis of a butyl analog of barbital, which was finally synthesized by Arthur Dox (Parke Davis and Company, Detroit, USA) in 1922, and marketed the following year by Abbott Laboratories, under the name Neonal® (Sneader 1985). Butobarbital (butethal in the USA) was three times as strong as barbital and its period of action was much shorter due to its lipophilicity, which greatly lowered the possibility of “rebound” drowsiness the day after administration.
In the years that followed, new barbiturates continued to come onto the market. In 1923, it was amobarbital (Amytal®), synthesized by Shonle and Moment (Eli Lilly Company, Indianapolis, USA) by adding a carbon atom to the butyl chain of butobarbital; and in 1929, Horace A Shonle also synthesized secobarbital (Seconal®). Both barbiturates had quite similar pharmacological properties to those of butobarbital (Sneader 1985). The next drugs of this series to be introduced were pentobarbital (Nembutal®), synthesized by Volwiler and Tabern (Abbott Laboratories) in 1930, and thiopental (Pentothal®). The latter, a sulfur derivative of pentobarbital, presented at the American Chemical Society congress in San Francisco in August 1935 (Tabern and Volwiler 1935), would revolutionize intravenous anesthesia and would be the only representative of the thiobarbiturate family to be officially recognized, being accepted first by the British Pharmacopoeia (1942, 7th Add) and subsequently by the United States Pharmacopoeia (1947, USP XIII) and the Pharmacopoeia Internationalis (1951, Volume I). Figure 3b shows an advertisement for pentobarbital in an American journal of the time.
Table 1 shows the recommended dosages of barbiturates used as hypnotics together with those of other drugs also used as hypnotics prior to the clinical introduction of benzodiazepines at the end of the 1950s. Among these last agents, chemically different from barbiturates although with similar pharmacological actions, we have to mention glutethimide (USV Pharmaceutical Corporation, 1954), methyprylon (Hoffmann-La Roche, 1955), methaqualone (King George Medical College, Lucknow, India, 1956; William H Rorer Inc, 1965), chlormethiazole (Hoffmann-La Roche, 1956), and ethchlorvynol (Pfizer, 1956). Most of these drugs were introduced as barbiturate substitutes, due to the fact that they seemed to offer a wider margin of safety. However, the clinical experience has demonstrated that their addiction liability and the severity of withdrawal symptoms were similar to those of barbiturates, and most of them were removed from the market some years later.
Table 1.
Mean and maximum dosage of the pharmacological agents used as hypnotics before the benzodiazepine era
| Dosage per administration | Daily maximum dosage | ||
|---|---|---|---|
| Drug | Mean dosage | Maximum dosage | |
| Ethchlorvynol | 250 mg | 500 mg | 750 mg |
| Chloral hydrate | 500 mg | 1000 mg | 1000 mg |
| Paraldehyde | 3 mL | 8 mL | 8 mL |
| Glutethimide | 250 mg | 500 mg | 500 mg |
| Methyprylon | 200 mg | 400 mg | 400 mg |
| Methaqualone | 200 mg | 400 mg | 600 mg |
| Phenobarbital | 50–100 mg | 200 mg | 200 mg |
| Amobarbital | 50–100 mg | 200 mg | 200 mg |
| Secobarbital | 100 mg | 200 mg | 200 mg |
| Pentobarbital | 100 mg | 200 mg | 200 mg |
| Sodium tripental | 250 mg | 500–1000 mg | – |
NOTE: The doses indicated correspond only to the hypnotic use of these drugs. The maximum doses of the barbiturates are not considered when they are used as anticonvulsants.
The role of barbiturates in “sleep cures” for schizophrenic patients
The hypnotic properties of some barbiturates were rapidly applied to the treatment of psychotic patients, thanks to their induction of a state of deep and prolonged sleep. The pioneer of these techniques was the Italian psychiatrist Giuseppe Epifanio, working at the University Psychiatric Clinic in Turin, who described his technique in an article published in 1915. The lack of impact of this development on the international scientific community can be attributed to the fact that it was published only in an Italian journal, and in the middle of the Great War (Epifanio 1915). It was on 25th March 1913 that Epifanio administered the first dose of Luminal to a girl aged 19 (FL) affected by manic-depressive psychosis, extending the treatment over a period of 4 days. The patient fell into a “deep sleep” that lasted until 9th April, was discharged at the end of June, and was in remission during the next two years. This case marked the beginning of what Manfred Bleuler would describe in 1955 as “the first of the great physical therapies” for mental disorders (Windholz and Witherspoon 1993).
However, the clinical introduction of these techniques is historically associated with Jakob Klaesi, a psychiatrist at the University Psychiatric Clinic in Zurich (Psychiatrische Universitätsklinik, Burghölzli, Switzerland). His “sleep cures” (“Dauerschlaf”, “Dauernarkose”), proposed in 1920 within the framework of the 59th Assembly of the Swiss Psychiatry Society (28th November 1920), enjoyed great prestige at the time and directly involved the use of barbiturates. Klaesi’s initial proposal was that his techniques for inducing deep hypnosis, taken from Epifanio, would facilitate communication between patient and psychotherapist (“to achieve a better relationship between doctor and patient”) (Shorter 1997, p 204). Klaesi introduced his method in Switzerland, and based it on pre-medication with morphine (0.01 mL) and scopolamine (0.001 mL) and the subsequent administration (intravenous or subcutaneous), over at least 6–7 days, of Somnifen® (Figure 4), a mixture of diethyl and dipropenyl-barbituric acid and diethylamine (2–4 mL), manufactured by the Hoffmann-LaRoche company. The percentage improvements reported by Klaesi, in samples of schizophrenic patients, ranged from 25% to 33%, which is 10% higher than the rates of spontaneous remission in this type of patient (Klaesi 1922). These cures (“prolonged sleep therapy”) acquired great popularity during the 1920s, with numerous variations as regards methodology and applications (agitated schizophrenic patients, delirium tremens, autism, morphine dehabituation, etc), though the administration of Somnifen was always involved (Windholz and Witherspoon 1993). Nevertheless, it is important to consider a fact mentioned in the first publication on the effectiveness of the method in schizophrenic patients: three of the 26 patients recruited died during the study due to bronchopneumonia or hemorrhages in the cardiac muscles (Klaesi 1922). A few years later, some authors set the mortality rate with Somnifen at around 5% (Müller 1927).
Figure 4.

The packaging of Somnifen®, produced by Hoffmann-LaRoche.
The legacy of Somnifen was taken up at the same Swiss clinic in Burghölzli by pharmacologist Max Cloetta and psychiatrist Hans W Maier, who sought a compound that would be better tolerated. In 1934, they prepared a compound based on paraldehyde, amylen hydrate, chloral hydrate, alcohol, ephedrine hydrate, digalen, and isopropyl-allyl-barbituric acid, which they called Cloettal® or “Cloetta Mixture”, and which was rectally administered (Cloetta and Meier 1934). This preparation was widely used in schizophrenic patients, not only in the Zurich clinic (Boss, Monnier), but also elsewhere, such as in the Soviet Union by Ivan P Pavlov (Windholz and Witherspoon 1993). The most rigorous study with this mixture was carried out in Burghölzli by Marcel Monnier, who, with a sample of 125 schizophrenic patients, applied strict exclusion criteria (elderly patients and those with renal or respiratory disorders) before applying the preparation. Only 84 patients were given the Cloetta Mixture, and 53 of them improved (40 were even discharged from the hospital). Nevertheless, two patients died during the treatment as a result of respiratory complications associated with the medication (Monnier 1936).
Eliot Slater, of the Maudsley Hospital in London, recalled that “sleep cures” were “the only treatment we had back in the 1930s that was of any value in acute psychotic disorders” (Slater 1975, p 74). After this initial period, the use of “sleep cures” based on barbiturates began to decline due in part to problems of safety, as well as to the clinical introduction of new biological therapies for the treatment of schizophrenic patients such as Sakel’s (1935) insulin shocks or the cardiazolic shocks of von Meduna (1937). Even so, as Shorter (1997) points out, “the story of barbituric narcosis has a corollary”. This refers to the work of D Ewen Cameron in the mid-1950s at the Psychiatry Department of the Allan Memorial Institute in Montreal (Canada). Financed by the Central Intelligence Agency (CIA), Cameron developed his technique of “psychic driving” (Cameron 1956), a prototype version of what would come to be known commonly as “brainwashing”. With this technique, in which barbiturates were also used, Cameron intended to take advantage of prolonged sleep to force his patients to listen to propaganda messages, which, in this case, were designed to quicken their recovery. In spite of its aims, eminently clinical, this work was widely criticized in the mass media at the time.
Barbiturates as antiepileptic agents
With phenobarbital, in addition to confirmation of the excellent hypnotic effect of barbiturates, it was demonstrated that these drugs had significant anticonvulsant properties. The discovery of these properties took place in 1912, the year of their commercialization, and provided another example of serendipity in the field of psychopharmacology. Alfred Hauptmann, resident psychiatrist in Freiburg, was given responsibility for the care of epileptic inpatients. Finding it impossible to sleep properly because of the continual convulsive seizures of his patients, Hauptmann decided to administer them some of the new hypnotics on the market, among them phenobarbital. Surprisingly, Hauptmann observed that the incidence of seizures in patients treated with low doses of phenobarbital fell notably, not only during the night, but also during the day (Hauptmann 1912). One of Hauptmann’s most important conclusions was that phenobarbital not only reduced the number of seizures, but also their intensity, allowing many patients to leave the institutions and enjoy a normal working life.
It was in this way that the anticonvulsant properties of barbiturates were discovered, phenobarbital being the first truly effective drug for the treatment of epilepsy (Iváñez and Díez-Tejedor 1998). Table 2 shows, by way of example, the anticonvulsant agents commonly employed in the treatment of epilepsy before and after the introduction of phenobarbital.
Table 2.
Anticonvulsant drugs used at the National Hospital (Queen Square) in London, before and after the clinical introduction of phenobarbital in the treatment of epilepsy
| 1910 | 1930 | ||
|---|---|---|---|
| Drugs of definite benefit | Drugs of doubtful benefit | Drugs of definite benefit | Drugs of doubtful benefit |
| Bromides | Monobromate of camphor | Bromides | Zinc salts |
| Chloral hydrate | Eosinate of sodium | Bromide combinations | Iron |
| Glycerophosphates | Chloretone | Phenobarbital | Digitalis |
| Borax | Antipyrin | Borax | Strophanthus |
| Belladonna | Double tartrate of | Calcium | |
| Zinc salts | borax and potassium | Opiates | |
| Opium | Belladonna | Hypnotics | |
| Strychnine | Nitroglycerine | ||
| Chloride of calcium | |||
| Atropine | |||
Adapted from Shorvon and Sander (1996).
However, the international acceptence of phenobarbital as an antiepileptic drug was seriously delayed, due first of all to the scarce significance outside Germany of the journal in which Hauptmann published the reports of his work (Münchener Medizinische Wochenschrift), and secondly, to the outbreak of World War I. Indeed, phenobarbitone was not commercialized in Great Britain until 1923, by the Winthrop Chemical Company. In one of his first reports on the use of phenobarbitone in England, Charles Brooks, Colony Medical Officer at the Chalfont Centre in London, noted its particular efficacy in severe cases of convulsions and in epileptic conditions with associated mental deficiency. Brooks also mentioned that if the barbiturate did not show a certain degree of effectiveness in the first months of treatment, the result of the therapy would not be satisfactory, so that it would be necessary to find an alternative (Brooks 1922). In a later report, Brooks made a close examination of patterns of use of phenobarbitone, concluding that it was more effective than bromides, but that it was not particularly useful in patients with low-intensity seizures (Brooks 1923).
It was precisely the Chalfont Centre that published, at the end of the 1920s, one of the first therapeutic guides for newly admitted epileptic patients, written by F Haward (Shorvon and Sander 1996). According to this guide, potassium bromide was the first-choice treatment, though it should be substituted by phenobarbital if there was no remission in the seizures within a given period of time (Table 2). If after three months of treatment the improvement was not clear, the guide recommended treatment with a combination of Luminal® and potassium bromide. Moreover, it set down the recommended dosage for phenobarbitone: 1 grain (65 grams) in the morning and another at night for adult patients, and 1/2 grain in the case of children; the dose was to be increased gradually, according to the clinical response, but should never exceed 6 grains per day (Haward 1928). At the beginning of the 1930s, the use of phenobarbital superseded definitively that of bromides in the treatment of epileptic seizures, despite the first reports of pharmacological tolerance and the risk of seizures when withdrawal was too abrupt. Phenobarbital is currently the most widely-prescribed antiepileptic drug in the world (Shorvon 2000), even though in the developed countries it has passed onto a secondary plane in therapy, for the treatment of partial and generalized seizures, due to its profile of adverse effects.
In the years following the discovery of the antiepileptic properties of phenobarbital, there were studies of numerous barbiturate derivatives in the field of epilepsy, the most important being mephobarbital (Prominal®) (Weese 1932) and, above all, deoxybarbital or primidone (Mysoline®). Primidone was synthesized by Bogue and Carrington (Imperial Chemical Industries Ltd, ICI, Manchester, UK) in 1949, demonstrating its antiepileptic activity in patients with generalized seizures in 1952 (Handley and Stewart 1952). Initially, primidone awoke great therapeutic interest, as it was thought that its anticonvulsant effectiveness may be greater than that of other available barbiturates, and without sedative effects (Bogue and Carrington 1953), but this interest soon waned after it was demonstrated that phenobarbital was a metabolite of this drug, together with phenyl-ethyl-malonamide (Butler and Waddell 1956). Comparative clinical studies carried out with phenobarbital and its prodrug, primidone, showed no differences between the two (Oleson and Dam 1967). Currently, primidone is still considered as being of some use in partial and secondary generalized seizures, but is not a first-choice drug. Unlike phenobarbital, it cannot be used in epileptic status, since no galenic formulation has been developed for its parenteral administration.
The discovery by Houston Merritt and Tracy Putnam (Boston City Hospital, USA) in 1938 of the anticonvulsant properties of phenytoin (the first drug to show that an antiepileptic need not be a hypnotic), in 1944 of trimethadione, and in the late 1950s of carbamazepine, extended the spectrum of antiepileptic drugs, resulting in decreased use of barbiturates in these applications.
The use of barbiturates in intravenous anesthesia
Despite the existence of some publications on the use of Somnifen® as a general anesthetic as early as 1921 by the French anesthetist Daniel Bardet – who noted that his patients woke up very slowly and with serious headaches (Bardet 1921) – the first barbiturate to be used systematically in anesthesia was sodium sec-butyl-(2-bromo-allyl)-barbiturate (Pernocton®). This was introduced into the field by the German obstetrician Bumm in 1927 (Bumm 1927). Subsequently, as new barbiturates were synthesized for their oral administration as sedatives, sodium salts of the same drugs were formulated, which could be administered intravenously and used as anesthetics (Dundee and McIlroy 1982). Notable among the pioneers in this field is John S Lundy of the Mayo Clinic (Rochester, USA), who introduced sodium amobarbital (1929) and sodium pentobarbital (1930) in anesthesia.
The addition of a methyl group to the butobarbital molecule, by the chemists Kropp and Taub at Bayer (IG Farbenindustrie, Leverkusen) in the early 1930s, gave rise to hexobarbital, whose sodium salt (Evipal®), introduced into clinical anesthesia in 1932 (Weese and Scharpff 1932), constituted the first barbiturate agent that induced anesthesia. Ten years after its introduction, more than 10 million people had undergone operations with the help of this drug (Adams 1944). The duration of hexobarbital’s action was shorter than that of its predecessors, given its greater lipophilicity, but under its effect some muscular movements occurred. This problem was solved through the next modification of the chemical structure of the basic nucleus of the barbiturates, the addition of a sulfur group to pentobarbital. Thus born were the agents that would revolutionize intravenous anesthesia, the thiobarbiturates, thanks to the work of Volwiler and Tabern of Abbott Laboratories (Tabern and Volwiler 1935). These agents were studied as anesthetics at the Mayo Foundation (Rochester) by John Lundy’s group, who gave the sulfur derivative of pentobarbital the name Thionembutal®. Its sodium salt was marketed as Pentothal (Figure 5). The team led by Ralph M Waters at the University of Wisconsin Medical School (Madison, USA) were the first to begin clinical administration of Pentothal, and published their results in 1936 (Pratt et al 1936). This agent rapidly displaced the rest of the barbiturates as an anesthetic, partly due to the swiftness of its onset and its short action period, and it currently remains the preferred intravenous anesthetic in many types of surgical intervention. Despite the anesthetic efficacy of both hexobarbital and thiopental, the barbiturates most commonly employed in surgery in the mid-20th century, they were not without their clinical problems. Such problems were brought to the public eye in particularly unfortunate fashion after the involvement of these agents, apparently due to malpractice, in numerous cases of death in patients treated in states of shock after the Japanese attack on Pearl Harbor in December 1941. Some authors went as far as describing these drugs as providing the “ideal form of euthanasia” (Halford 1943).
Figure 5.
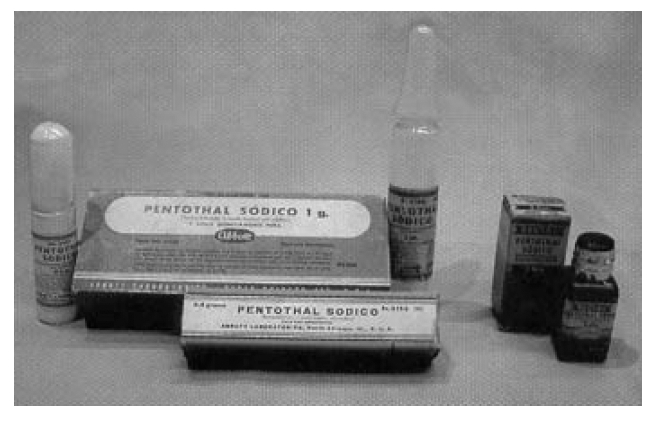
The packaging of Abbott Pentothal® at the time of its clinical introduction in the late 1930s. Pieces from the Museum of the Buenos Aires Anaesthesiology Association (Argentina).
After World War II the search for anesthetic barbiturates continued, and new compounds such as thiobutobarbital (Horatz and Stürtzbecher 1952) were introduced, though the only one that truly challenged thiopental was methohexital (Brietal®), developed by SM Chernish’s group at Lilly Research Laboratories (Indianapolis, USA) in 1956. In clinical trials, methohexital showed itself to be more potent than thiopental and to lead to quicker recovery in patients; it was recommended for use as an anesthetic inducer in minor outpatient surgery (Taylor and Stoelting 1960). The subsequent development of other anesthetic agents for intravenous administration (hydroxydione, alphaxalone, etomidate, propofol, etc) led to a reduction in the use of barbiturates in this context.
The peak and decline of barbiturate therapy
As mentioned earlier, chemists from different universities and pharmaceutical companies managed to synthesize over 2500 barbiturate derivates. The differential pharmacokinetic properties of these agents made it possible to draw up a practical clinical classification, based on the duration of their pharmacological action (Hollister 1983). Thus, the barbiturates in the category of short or intermediate action (secobarbital, amobarbital, pentobarbital) were employed initially as hypnotics, whilst those of prolonged action (phenobarbital) were widely used as anxiolytics and anticonvulsants; ultrashort-acting agents, notably sodium thiopental, were especially useful as anesthetic inducers for minor operations (Table 3). From time to time, some barbiturates have been used in the treatment of other disorders. One such case is the use of primidone in the management of essential tremor (Koller et al 2000), while another is that of combinations of barbiturates and analgesics (salicylates, codeine, etc) in the treatment of headaches, migraines, and other types of pain (Wolf et al 1941), though such applications are considered counterproductive today.
Table 3.
Classification and principal clinical applications of the barbiturates most commonly employed before World War II
| Barbiturates | Trade name | Chemical name | Clinical indications | |
|---|---|---|---|---|
| Long-acting | Phenobarbital | Luminal | 5-ethyl-5-phenylbarbituric acid | Sedative |
| Intermediate-acting | Amobarbital | Amytal | 5-ethyl-5-isopentylbarbituric acid | Hypnotic |
| Short-acting | Pentobarbital | Nembutal | 5-ethyl-5-(1-methylbutyl)-barbituric acid | Hypnotic and anticonvulsant |
| Secobarbital | Seconal | 5-allyl-5-(1-methylbutyl)-barbituric acid | Hypnotic | |
| Ultrashort-acting | Thiopental | Pentothal | 5-ethyl-5-(1-methylbutyl)-thiobarbituric acid | Anesthesia inducer |
Adapted from Hollister (1983).
Some barbiturates, such as sodium amytal and sodium pentothal (the latter being known as “the truth serum”) were widely known and used as coadjuvant agents for the exercise of narcoanalysis, as initially developed by Bleckwenn in 1930 (Bleckwenn 1930a, 1930b). In principle, the application of an infusion of barbiturates reverted temporarily the catatonic state of certain schizophrenic patients. These cures for catatonia allowed patients, for a few hours, to maintain conversations and interact with their environment, before returning to their state of lethargy. Despite the fact that the response was somewhat brief, these cures were quite customary in European asylums in the 1930s and 1940s. But a variety of this technique became widespread during and after World War II: it consisted of the intravenous administration of a short-acting barbiturate, which had a disinhibiting effect (potentiating positive transfers) and facilitated the subsequent exercise of psychotherapy (a phenomenon referred to as “cathartic abreaction”) (Lehmann 1993). This technique was also called by other authors the “induced crepuscular method”.
It was during the 1930s and 1940s that barbiturates attained their greatest popularity and were most widely used, putting them in a position that could be compared, according to Hollister (1983), to that currently held by benzodiazepines. The barbiturates most commonly used at that time were phenobarbital, sodium amobarbital, sodium secobarbital, sodium pentobarbital, and sodium thiopental. Despite their widespread use during the first half of the 20th century, no barbiturate succeeded in eliminating the main drawbacks of these drugs, which were the phenomena of dependence and death by overdose (Johns 1977). Among the paradoxes of destiny is the possible death through overdose of the two scientists who introduced the first barbiturate, Fischer and von Mering, after some years of dependence upon these substances (Escohotado 1996). To reduce these problems, from a legal perspective, a series of laws were passed aimed at regulating the distribution and sale of barbiturates. The first of these came into force in California in 1929. However, its effects were limited, if we consider, for example, that the production of barbiturates in the USA increased by more than 400% from 1933, with some 70 tons of these drugs sold in 1936. The problem continued during the following decade, and it became necessary to arrange special conferences for all those involved, such as that held in Washington, under the auspices of the American Pharmaceutical Association, on 12th October 1945 (Conference on the Regulation of Use and Distribution of Barbiturates). Barbiturate use in the pre-benzodiazepine period was such that, in the USA alone, production of these drugs reached, in 1955, the quantity necessary for the treatment of 10 million people throughout an entire year. Figure 6 shows the industrial production of barbiturates and their derivatives in the USA during the 1940s and 1950s.
Figure 6.
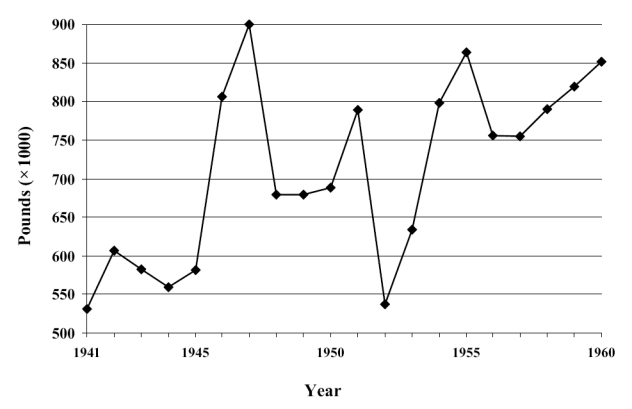
Evolution of annual barbiturates production in USA for the period 1941–1960. Adapted from Fort (1964).
The capacity of barbiturates to cause dependence was described in the medical literature as early as one year after the commercialization of barbital (“the Veronal habit”), though reliable evidence of the potential of these drugs to generate abuse was not available until the 1950s (Glatt 1962). In fact, doses 4–6 times higher than the therapeutic dose as hypnotics of the short-acting barbiturates (400–600 mg/day of amobarbital, secobarbital, or pentobarbital) brought about, if the treatment was sufficiently prolonged, authentic withdrawal syndromes when use was stopped. In order to palliate these effects, the Narcotics Expert Committee at the World Health Organization recommended (at their sessions of 7th–12th January, 1952, and 18th–24th October, 1956) that barbiturates should only be available on medical prescription. In spite of this, and according to different estimates, in 1965 there were 135 000 barbiturate addicts in England, whilst in the United States it was declared, by a special drug-dependence committee set up by President Kennedy in 1962, that there may be as many as 250 000 Americans addicted to barbiturates. Indeed, the USA currently produces 30 barbiturate pills per inhabitant per year (Escohotado 1996). Some barbiturates (amobarbital and pentobarbital) have even found their way into mixtures with amphetamine derivatives (goofballs), such as Dexamyl®, a combination of dextroamphetamine and amobarbital.
In relation to the frequent cases of death by overdose, given the small therapeutic margin of these substances, it should be pointed out that this was a common method in suicide attempts. It suffices to recall, in this regard, the famous case of Marilyn Monroe, on whose death certificate it clearly states “acute poisoning by overdose of barbiturates” (Figure 7). The lethal effect of these compounds was such that a mixture of barbiturates with other substances was even employed in some USA states for the execution of prisoners sentenced to death. Furthermore, there are classic reports of fatal overdose due to the “automatism phenomenon”, whereby the patient would take his or her dose, only to forget that he or she had already taken it, given the amnesic effect of the drug, and take it again, this process being repeated several times (Richards 1934). Figure 8 shows the evolution of number of deaths (accidental or suicide) by barbiturate overdose in England and Wales for the period 1905–1960. In this regard, and in the city of New York alone, in the period 1957–1963, there were 8469 cases of barbiturate overdose, with 1165 deaths (Sharpless 1970), whilst in the United Kingdom, between 1965 and 1970, there were 12 354 deaths attributed directly to barbiturates (Barraclough 1974). These data should not surprise us, since in a period of just one year (1968), 24.7 million prescriptions for barbiturates were issued in the United Kingdom (Plant 1981). In view of these data, the Advisory Council Campaign in Britain took measures restricting the prescription of these drugs. Meanwhile, the prescription of prolonged-acting sedative barbiturates was strongly opposed through citizens’ action campaigns such as CURB (Campaign on the Use and Restrictions of Barbiturates), especially active during the 1970s.
Figure 7.

Death certificate of the actress Marilyn Monroe, issued on 28th August 1962. The circles indicate cause of death (“Acute barbiturate poisoning. Ingestion of overdose”) and the intentionality (“Probable suicide”)
Figure 8.

Deaths from overdose of barbiturates in England and Wales during the period 1905–1960 (Registrar-General’s Statistical Review for England and Wales). Includes both accidental deaths and suicides. Adapted from Glatt (1962).
Furthermore, during the 1950s, when the use of barbiturates was at its peak, there took place a veritable revolution in the approach to psychiatric disorders, thanks to the introduction into clinical practice of the first pharmacological tools aimed specifically at treating these patients (Caldwell 1970; Jacobsen 1986; Ayd 1991; Lehmann 1993; Frankenburg 1994; López-Muñoz et al 2000; Ban 2001; Healy 2002). This “psychopharmacological revolution” began with the discovery and clinical use, from 1952, of chlorpromazine (López-Muñoz et al 2004), culminating in the commercialization of the first benzodiazepine, chlordiazepoxide, in 1960. The discovery of benzodiazepines was actually made possible, in part, by the 60 years of clinical and basic research provided by barbiturates, whose therapeutic life, from that time on, began to decline.
Barbiturates today
Currently, the use of barbiturates is circumscribed to quite specific therapeutic applications (Charney et al 2001). Thus, phenobarbital and butabarbital are still used as sedatives in cases of gastrointestinal and asthmatic functional disorders, as well as to antagonize the adverse central stimulant effects of some drugs, such as ephedrine, dextroamphetamine, or theophylline. Phenobarbital is also used in cases of withdrawal syndromes of hypnosedative agents. In the field of neurology, barbiturates (phenobarbital and primidone) are still employed, not only in the treatment of certain types of epilepsy (partial and tonic-clonic generalized seizures), but also in the emergency treatment of some types of convulsions, such as those associated with tetanus, eclampsia, cerebral hemorrhage, status epilepticus, or different forms of poisoning. As intravenous anesthetic inducers, ultrashort-acting barbiturates are of use, mainly thiopental and methohexital, the latter also being administered rectally in children or as a sedative in some diagnostic imaging explorations. Table 4 shows the therapeutic applications of barbiturates that have survived to the present day.
Table 4.
Barbiturates currently employed and therapeutic applications
| Barbiturate | Routes of administration | Therapeutic uses |
|---|---|---|
| Amobarbital | Oral, IM, IV | Insomnia |
| Preoperative sedation | ||
| Emergency management of seizures | ||
| Aprobarbital | Oral | Insomnia |
| Butabarbital | Oral | Insomnia |
| Preoperative sedation | ||
| Mephobarbital | Oral | Epilepsy |
| Daytime sedation | ||
| Methohexital | IV | Induction/maintenance of anesthesia |
| Pentobarbital | Oral, rectal, IM, IV | Insomnia |
| Preoperative sedation | ||
| Emergency management of seizures | ||
| Phenobarbital | Oral, IM, IV | Epilepsy |
| Status epilepticus | ||
| Daytime sedation | ||
| Primidone | Oral | Epilepsy |
| Secobarbital | Oral, rectal, IM, IV | Insomnia |
| Preoperative sedation | ||
| Emergency management of seizures | ||
| Thiopental | Rectal, IV | Induction/maintenance of anesthesia |
| Preoperative sedation | ||
| Emergency management of seizures |
Adapted from Charney et al (2001).
Abbreviations: IM, intramuscular; IV, intravenous.
In addition to these approved indications, the barbiturates present other current uses. Phenobarbital is capable of improving the hepatic transport of bilirubin in patients with hemolytic jaundice, so that it can be used in newborn babies to treat hyperbilirubinemia and kernicterus. At a diagnostic level, amobarbital, in low doses, can be injected directly into the carotid artery prior to neurosurgery to identify the dominant cerebral hemisphere. Finally, anesthetic doses of barbiturates can attenuate post-surgical cerebral edemas and have positive effects in cases of cardiac and cerebral ischemia, reducing the size of the infarcted region. Moreover, barbiturates have been used since the 1970s in the management of acute traumatic brain injury in their capacity to reduce intracranial pressure (Marshall et al 1979). The mechanism through which high-dose barbiturates appear to exert their intracranial pressure-lowering effects is double: reduction of metabolism (with the consequent lower oxygen demand by cerebral tissue) and modifications in vascular tone (Kassell et al 1980). Additionally some direct neuroprotective effects, such as membrane stabilization or inhibition of free radical-mediated lipid peroxidation, have been postulated (Piatt and Schiff 1984). Despite results of the multicenter randomized clinical trial published by Eisenberg et al (1988) that demonstrated the efficacy of high-dose barbiturates in severely head-injured patients with intractable intracranial pressure elevations, recent collaborations, based in Cochrane methodology, concluded that there is no evidence of health improvement in this type of patient (Roberts 2000).
The barbiturates introduced clinically one century ago were the first pharmacological agents to have demonstrated – in an historical period that was therapeutically inhospitable – a real efficacy in different neuropsychiatric disorders. They were the first-line treatment as hypnotics and anticonvulsants during the first half of the 20th century. The clinical results obtained in the last years in other indications such as the treatment (acute or prophylactic) of traumatic brain injury, although contradictory, seems to confirm that, from the pharmacological perspective, the barbiturates continue furnishing certain novelties and that in their history the last page has not yet been written.
References
- Adams RC. Intravenous anesthesia. New York: Hoeber; 1944. [Google Scholar]
- Alamo C, López-Muñoz F, Echániz T, et al. Fármacos ansiolíticos, sedantes e hipnóticos. In: López-Muñoz F, Alamo C, editors. Historia de la Neuropsicofarmacología. Una nueva aportación a la terapéutica farmacológica de los trastornos del Sistema Nervioso Central. Madrid: Ediciones Eurobook SL; 1998. pp. 245–68. [Google Scholar]
- Ayd FJ. The early history of modern psychopharmacology. Neuropsychopharmacology. 1991;5:71–84. [PubMed] [Google Scholar]
- Balme R. Early medicinal uses of bromides. J Royal Coll Physic. 1976;10:205–8. [PMC free article] [PubMed] [Google Scholar]
- Ban TA. Pharmacotherapy of mental illness. A historical analysis. Prog Neuro-Psychopharmacol Biol Psychiatr. 2001;25:709–27. doi: 10.1016/s0278-5846(01)00160-9. [DOI] [PubMed] [Google Scholar]
- Bardet D. Sur l’utilisation. Comme anésthésique géneral, d’un produit nouveau, le diéthyl-diallyl-barbiturate de diéthylamine. Bull Gén Thèrap Mèd Chirurg Obstètr Pharm. 1921;172:27–33. [Google Scholar]
- Barraclough BM. Are there safer hypnotics than barbiturates. Lancet. 1974;i:57–8. doi: 10.1016/s0140-6736(74)93055-4. [DOI] [PubMed] [Google Scholar]
- Bleckwenn WJ. Narcosis as therapy in neuropsychiatric conditions. JAMA. 1930a;95:1168–71. [Google Scholar]
- Bleckwenn WJ. Production of sleep and rest in psychotic cases. Arch Neurol Psychiatry. 1930b;24:365–75. [Google Scholar]
- Bogue JY, Carrington HC. The evaluation of mysoline – a new anticonvulsant drug. Br J Pharmacol. 1953;8:230–5. doi: 10.1111/j.1476-5381.1953.tb00784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C The National Society for Epileptics. Thirtieth Annual Report. Report of the Medical Officer. 1922. p. 19. [Google Scholar]
- Brooks C The National Society for Epileptics. Thirty-first Annual Report. Report of the Medical Officer. 1923. p. 23. [Google Scholar]
- Bumm R. Intavenose Narkosen mit Barbitur-saurederivaten. Klin Wochenschr. 1927;6:725–6. [Google Scholar]
- Butler TC, Waddell WJ. Metabolic conversion of primidone (Mysoline) to phenobarbital. Proc Soc Exp Biol NY. 1956;93:544–66. doi: 10.3181/00379727-93-22813. [DOI] [PubMed] [Google Scholar]
- Caldwell AE. History of psychopharmacology. In: Clark WG, Del Giudice J, editors. Principles of psychopharmacology. New York: Academic Pr; 1970. pp. 9–30. [Google Scholar]
- Cameron DE. Psychic driving. Am J Psychiatry. 1956;112:502–9. doi: 10.1176/ajp.112.7.502. [DOI] [PubMed] [Google Scholar]
- Carter MK. The history of barbituric acid. J Chem Educ. 1951;28:525–8. [Google Scholar]
- Cervello V. Sull’ azione fisiologica della paraldeide e contributio allo studio del cloralio idrato. Ricerche. Arch Soc Med. 1882;6:177–214. [Google Scholar]
- Charney DS, Mihic SJ, Harris RA. Hypnotics and sedatives. In: Hardman JG, Limbird LE, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 10. New York: MacGraw-Hill; 2001. pp. 399–427. [Google Scholar]
- Chernish SM, Gruber CM, Demeyer M, et al. Double blind comparison of compound 22451, pentothal and surital. Fed Proc. 1956;15:409. [Google Scholar]
- Cloetta M, Maier AW. Über eine Verbesserung der psychiatrischen Dauernarkosebehandlung. Zeitsch gesamte Neurol Psychiatrie. 1934;164:146–62. [Google Scholar]
- Cohen WAT. Chemisch-Historische Aanteekeningen. De nomenclatuur van enkele organische zuren. Chemisch Weekblad. 1943;40:176. [Google Scholar]
- Conrad M, Guthzeit M. Über Barbitur-saurederivate. Berichte. 1881;14:1943. [Google Scholar]
- Dundee JW, McIlroy PDA. The history of the barbiturates. Anaesthesia. 1982;37:726–34. doi: 10.1111/j.1365-2044.1982.tb01310.x. [DOI] [PubMed] [Google Scholar]
- Eisenberg HM, Frankowski RF, Contant CF, et al. High-dose barbiturate control of elevated intracranial pressure in patient with severe head injury. J Neurosurg. 1988;69:15–23. doi: 10.3171/jns.1988.69.1.0015. [DOI] [PubMed] [Google Scholar]
- Epifanio G. L’ipnosi farmacologica prolungata e sua applicazione per la cura di alcune psicopatici. Riv Patol Nerv Mentale. 1915;20:273–308. [Google Scholar]
- Escohotado A. Historia elemental de las drogas. Barcelona: Editorial Anagrama; 1996. [Google Scholar]
- Fieser LF. Organic chemistry. Boston: DC Heath and Company; 1944. p. 247. [Google Scholar]
- Fischer E, von Mering J. Ueber ein neue Klasse von Schlafmitteln. Therapie Gegenwart. 1903;44:97–101. [Google Scholar]
- Fort J. The problem of barbiturates in the United States of America. UNDOC Bull Narc. 1964;1:17–35. [Google Scholar]
- Frankenburg FR. History of the development of antipsychotic medication. Psychiatr Clin North Am. 1994;17:531–40. [PubMed] [Google Scholar]
- Glatt MM. The abuse of barbiturates in the United Kingdom. UNODC Bull Narc. 1962;2:19–38. [Google Scholar]
- Griesinger W. Die Pathologie und Therapie der psychischen Krankheiten. 2. Stuttgart: Krabbe; 1861. [Google Scholar]
- Grimaux E. Synthese des dérivés uriques de la série de l’alloxane. Bull Soc Chim France. 1879;31:146. [Google Scholar]
- Halford FJ. A critique of intravenous anesthesia in war surgery. Anesthesiology. 1943;4:24–30. [Google Scholar]
- Handley R, Stewart ASR. Mysoline: a new drug in the treatment of epilepsy. Lancet. 1952;262:742. doi: 10.1016/s0140-6736(52)90500-x. [DOI] [PubMed] [Google Scholar]
- Hauptmann A. Luminal bei Epilepsie. MÜnch Med Wochenschr. 1912;59:1907. [Google Scholar]
- Haward FC The National Society for Epileptics. Thirty-fifth Annual Report. Report of the Medical Officer. 1928. p. 24. [Google Scholar]
- Healy D. The creation of psychopharmacology. Cambridge: Harvard Univ Pr; 2002. [Google Scholar]
- Hollister LE. The pre-benzodiazepine era. J Psychoactive Drugs. 1983;15:9–13. doi: 10.1080/02791072.1983.10472117. [DOI] [PubMed] [Google Scholar]
- Horatz K, StÜrtzbecher F. Neue Hilfsmittel in der Anaesthesie. Anaesthesist. 1952;1:149–50. [PubMed] [Google Scholar]
- Impens E. Pharmakologisches über Luminal, oder Phenylethil barbiturat-saure, ein neues Hypnoticum. Dtsch Med Wochenschr. 1912;38:945–7. [Google Scholar]
- Iváñez V, Díez-Tejedor E. Fármacos antiepilépticos y anticonvulsivantes: aspectos históricos. In: López-Muñoz F, Alamo C, editors. Historia de la Neuropsicofarmacología. Una nueva aportación a la terapéutica farmacológica de los trastornos del Sistema Nervioso Central. Madrid: Ediciones Eurobook SL; 1998. pp. 347–64. [Google Scholar]
- Jacobsen E. The early history of psychotherapeutic drugs. Psychopharmacology. 1986;89:138–44. doi: 10.1007/BF00310617. [DOI] [PubMed] [Google Scholar]
- Johns MW. Self-poisoning with barbiturates in England and Wales during 1959-74. BMJ. 1977;i:1128–30. doi: 10.1136/bmj.1.6069.1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliusburger O. Ueber Luminal einneues Hypnoticum und Sedativum. Berl Klin Wochenschr. 1912;49:940–2. [Google Scholar]
- Kassell NF, Hitchon PW, Gerk MK, et al. Alterations in cerebral blood flow, oxygen metabolism, and electrical activity produced by high-dose thiopental. Neurosurgery. 1980;7:598–603. doi: 10.1227/00006123-198012000-00011. [DOI] [PubMed] [Google Scholar]
- Kast A. Sulfonal, ein Neues Schlafmittel. Berl Klin Wochenschr. 1888;25:309–14. [Google Scholar]
- Klaesi J. Über die therapeutische Anwendung der “Dauernarkose” mittels Somnifens bei Schizophrenen. Zeitsch gesamte Neurol Psychiatrie. 1922;74:557–92. [Google Scholar]
- Koller WC, Hristova A, Brin M. Pharmacological treatment of essential tremor. Neurology. 2000;54(11 Suppl 4):30–8. [PubMed] [Google Scholar]
- Lehmann HE. Before they called it psychopharmacology. Neuropsychopharmacology. 1993;8:291–303. doi: 10.1038/npp.1993.69. [DOI] [PubMed] [Google Scholar]
- Lehmann HE, Ban TA. Pharmacotherapy of tension and anxiety. Springfield: Charles C Thomas; 1970. [PubMed] [Google Scholar]
- Liebreich O. Eine Arzneimittel-Untersuchung. Berlin: Muller; 1869. Das Chloralhydrat ein neues Hypnoticum und Anästheticum, und dessen Anwendung in die Medizin. [Google Scholar]
- Locock C. Discussion of paper by EH Sieveking. Analysis of 52 cases of epilepsy observed by author. Lancet. 1857;i:527. [Google Scholar]
- Loewe S. Klinische Erfahrungen mit Luminal. Dtsch Med Wochenschr. 1912;38:947–8. [Google Scholar]
- López-Muñoz F, Alamo C, Cuenca E. La “Década de Oro” de la Psicofarmacología (1950-1960): Trascendencia histórica de la introducción clínica de los psicofármacos clásicos, Psiquiatria.COM (electronic journal), Sep, 4 (3), URL: http://www.psiquiatria.com/psiquiatria/revista/47/1800/?++interactivo. 2000
- López-Muñoz F, Alamo C, Rubio G, et al. Half a century since the clinical introduction of chlorpromazine and the birth of modern psychopharmacology. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28:205–8. doi: 10.1016/s0278-5846(03)00165-9. [DOI] [PubMed] [Google Scholar]
- Lundy JS. A case illustrating the use of sodium iso-amylethyl barbiturate (sodium amytal). Report of the 48th Meeting of the Society of the Clinical Surgery; Apr 29-30.1929. [Google Scholar]
- Lundy JS. Intravenous anesthesia: particularly hypnotic, anesthesia and toxic effects of certain new derivates of barbituric acid. Anesth Analg. 1930;9:210–17. [Google Scholar]
- Marshall LF, Smith RW, Shapiro HM. The outcome with aggressive treatment in severe head injuries: acute and chronic barbiturate administration in the management of head injury. J Neurosurg. 1979;50:26–30. doi: 10.3171/jns.1979.50.1.0026. [DOI] [PubMed] [Google Scholar]
- McLeod N. The hormone sleep: a new departure in the treatment of acute mania. BMJ. 1900;i:134–6. doi: 10.1136/bmj.1.2038.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrit HH, Putnam TJ. Sodium diphenylhydantoinate in treatment of convulsive disorders. JAMA. 1938;111:1068–73. [Google Scholar]
- Monnier M. Die Dauerschlafbehandlung der Schizophrenien mit Narkosenmischung von Cloetta an der Psychiatrischen Klinik Burghölzli – Zürich. Nervenartz. 1936;9:14–29. [Google Scholar]
- Müller M. Die Dauernarkose mit flussigem Dial bei Psychosen, speziell bei manisch-depressivem Irresein. Zeitsch gesamte Neurol Psychiatrie. 1927;107:522–43. [Google Scholar]
- Norton A. Depression. BMJ. 1979;2:429–30. doi: 10.1136/bmj.2.6187.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleson OV, Dam M. The metabolic conversion of primidone to phenobarbitone in patients under long-term treatment. Acta Neurol Scand. 1967;43:348–56. doi: 10.1111/j.1600-0404.1967.tb05737.x. [DOI] [PubMed] [Google Scholar]
- Piatt JH, Schiff SJ. High dose barbiturate therapy in neurosurgery and intensive care. Neurosurgery. 1984;15:427–44. doi: 10.1227/00006123-198409000-00023. [DOI] [PubMed] [Google Scholar]
- Plant M. What aetiologies? In: Edwards G, Busch C, editors. Drug problems in Britain. A review of ten years. London: Academic Pr; 1981. pp. 245–80. [Google Scholar]
- Pratt TW, Tatum AL, Hathaway HR, et al. Sodium ethyl (1-methyl butyl) thiobarbiturate: preliminary experimental and clinical study. Am J Surg. 1936;31:464–6. [Google Scholar]
- Richards R. Symptoms of poisoning bu hypnotics of barbituric acid groups. BMJ. 1934;i:331. [Google Scholar]
- Roberts I. Barbiturates for acute traumatic brain injury. Cochrane Database Syst Rev. 2000;2:CD000033. doi: 10.1002/14651858.CD000033. [DOI] [PubMed] [Google Scholar]
- Sakel M. Neue Behandlung der Schizophrenie. Vienna: Perles; 1935. [Google Scholar]
- Scott DF. J Hist Neurosci. Vol. 1. 1992. The discovery of antiepileptic drugs; pp. 111–18. [DOI] [PubMed] [Google Scholar]
- Sharpless SK. The barbiturates. In: Goodman LS, Gilman A, editors. The pharmacological basis of therapeutics. 4. New York: The MacMillan Company; 1970. pp. 98–120. [Google Scholar]
- Shonle HA, Moment A. Some new hypnotics of the barbituric acid series. J Am Chem Soc. 1923;45:243–9. [Google Scholar]
- Shorter E. A history of psychiatry. From the era of the asylum to the age of Prozac. New York: J Wiley; 1997. [Google Scholar]
- Shorvon SD. Handbook of epilepsy treatment. Oxford: Blackwell Science; 2000. [Google Scholar]
- Shorvon SD, Sander JWAS. Historical introduction. The treatment of epilepsy at the National Hospital; Queen Square, 1857-1939: a mirror of the first phase of the modern history of medical and surgical therapy. In: Shorvon SD, Dreifuss F, Fish D, et al., editors. The treatment of epilepsy. Oxford: Blackwell Science; 1996. pp. xvii–xliv. [Google Scholar]
- Slater E. Psychiatry in the thirties. Contemp Rev. 1975;226:70–5. [PubMed] [Google Scholar]
- Sneader W. Drug discovery: the evolution of modern medicines. Chichester: J Wiley; 1985. [Google Scholar]
- Sourkes TL. Early clinical neurochemistry of CNS-active drugs. Chloral hydrate. Mol Chem Neurophath. 1992;17:21–30. doi: 10.1007/BF03159978. [DOI] [PubMed] [Google Scholar]
- Tabern DL, Volwiler EH. Sulfur-contained barbiturate hypnotics. J Am Chem Soc. 1935;57:1961–3. [Google Scholar]
- Taylor C, Stoelting VK. Methohexital sodium – a new ultrashort acting barbiturate. Anesthesiology. 1960;21:29–34. [Google Scholar]
- Volwiler EH, Tabern DL. 5,5-substituted barbituric acid. J Am Chem Soc. 1930;52:393–407. [Google Scholar]
- Von Baeyer A. Untersuchungen über die Harnsauregruppe. Annalen. 1864;130:129. [Google Scholar]
- Von Husen H. Über Veronal. PNW. 1904;6:57–61. [Google Scholar]
- Von Meduna L. Die Konvulsionstherapie der Schizophrenie. Halle: Marhold; 1937. [Google Scholar]
- Weese H. Pharmakologie des Prominal. Dtsch Med Wochenschr. 1932;58:696. [Google Scholar]
- Weese H, Scharpff W. Evipan, ein neuartiges Einschlafmittel. Dtsch Med Wochenschr. 1932;58:1205–7. [Google Scholar]
- Windholz G, Witherspoon LH. Sleep as cure for schizophrenia: a historical episode. Hist Psychiatry. 1993;4:83–93. doi: 10.1177/0957154X9300401304. [DOI] [PubMed] [Google Scholar]
- Wolf HG, Hardy JD, Goodell H. Measurement of the effect of the pain threshold of acetylsalicylic acid, acetanilid, acetophenetidin, aminopyrine, ethyl alcohol, trichlorethylene, a barbiturate, quinine, ergotamine, tartrate and caffeine, and analysis of their relation to the pain experience. J Clin Invest. 1941;20:63–80. doi: 10.1172/JCI101196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward SB. Observations on the medical treatment of insanity (1846) Am J Psychiatry. 1994;151(Suppl 6):220–30. doi: 10.1176/ajp.151.6.220. [DOI] [PubMed] [Google Scholar]