

Abstract
Currently available genetic and archaeological evidence is generally interpreted as supportive of a recent single origin of modern humans in East Africa. However, this is where the near consensus on human settlement history ends, and considerable uncertainty clouds any more detailed aspect of human colonization history. Here, we present a dynamic genetic model of human settlement history coupled with explicit geographical distances from East Africa, the likely origin of modern humans. We search for the best-supported parameter space by fitting our analytical prediction to genetic data that are based on 52 human populations analyzed at 783 autosomal microsatellite markers. This framework allows us to jointly estimate the key parameters of the expansion of modern humans. Our best estimates suggest an initial expansion of modern humans ∼56,000 years ago from a small founding population of ∼1,000 effective individuals. Our model further points to high growth rates in newly colonized habitats. The general fit of the model with the data is excellent. This suggests that coupling analytical genetic models with explicit demography and geography provides a powerful tool for making inferences on human-settlement history.
The availability of a large data set of DNA samples from >1,000 individuals distributed worldwide and typed at hundreds of genetic markers1,2 has led to the description of extremely strong patterns in the geographic distribution of genetic diversity in humans. Genetic differentiation between populations increases essentially linearly with geographic distance, computed along landmasses.3–6 Even more striking is the observation that geographic distance along landmasses from East Africa (a likely origin of anatomically modern humans) is an excellent predictor of the genetic diversity of individual populations throughout the world. Indeed, genetic diversity decreases smoothly with increasing geographic distance from Africa.7,8
These patterns offer compelling evidence for the hypothesis of a recent African origin of modern humans.9–13 They further suggest a scenario of the colonization of the world by modern humans through a large number of successive bottlenecks of small amplitude and a predominance of gene flow over limited distance.7 The simple nature of the patterns described, their smoothness, and the large proportion of variance explained by linear regressions offer an exciting opportunity to model these patterns with tractable population-genetics models, to gain insight into the key parameters of human-settlement history.
In this article, we consider an analytical dynamic colonization population-genetics model in a one-dimensional habitat, to simulate the process of colonization of the world by modern humans and their population expansion. Although the colonization of the world by anatomically modern humans was obviously a two-dimensional process, our one-dimensional framework is appropriate because we consider only within-population coalescence times so that we do not have to make any assumption about the connection between populations. Using this framework, we simulate the evolution of coalescence times during the colonization process, to estimate parameter values that provide the best fit to the variance in allele size computed in 52 populations distributed worldwide. Specifically, we search for the values of the age of the initial expansion, the number of individuals at the source of this event, as well as growth rate and carrying capacity of subsequently colonized demes. Our simulations point to an expansion of modern humans ∼56,000 years ago, starting from an ancestral source population of ∼1,000 effective individuals, and also suggest high population-growth rates within newly colonized demes.
Material and Methods
General Features of the Model
To model the colonization of the world from a single ancestral African population, we take advantage of a framework developed by Austerlitz and colleagues.14 The model is based on a one-dimensional habitat with d demes arranged linearly (fig. 1). At the beginning of the colonization process, a single deme is occupied (which would correspond to the first modern human population that appeared in Africa). This first deme is located at one border of the one-dimensional habitat, and its size is at the carrying capacity K0. All the other demes have equal carrying capacity K.
Figure 1. .

Schematic representation of the colonization model. At generation t0, the entire population consists of a single ancestral deme at the border of the stepping-stone. This initial founding population has a carrying capacity K0 and is at mutation-drift equilibrium. At each generation, this population sends a proportion of m/2 migrants to the neighboring site (deme 2). Thus, at generation t1, deme 2 comprises (m/2)K0 individuals. The individuals in deme 2 reproduce randomly and produce a number of offspring determined by the growth rate parameter r. Once deme 2 has reached its carrying capacity K (at times tk), it starts sending migrants to its two neighboring sites. This process of migration and population growth of newly colonized demes continues until all demes are full.
Individuals are diploid and mate at random within demes, and generations are nonoverlapping. Every deme at carrying capacity sends an equal number of migrants (Km/2) to its two neighboring demes, where m is the migration rate. Demes are thus colonized sequentially (fig. 1). Reproduction occurs after the colonization/migration phase. We chose a logistic population-growth model to describe the evolution of population size in each deme, which is biologically more realistic than exponential growth or sudden expansion.11,15
From this demographic model, we can obtain the dynamics, over time, of the expected coalescence times of pairs of genes within and between demes. This can be expressed as a matrix T with elements ti,j, the expected coalescence time between genes sampled in deme i and deme j. We now express migration in matrix form, with Mt being a backward migration matrix adjusted for subpopulation size and with elements mi,j being the probability that a gene sampled in deme i at generation t originated from deme j in the previous generation. The recursion of coalescence times (eq. [13] in the work of Austerlitz et al.14) can be rewritten as
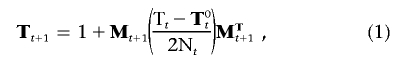 |
where T0t is a matrix with the same diagonal elements as Tt, but all other elements are set to zero. Equation (1) expresses the dynamics of expected coalescence times under different demographic scenarios. The initial conditions are given by T0, where all elements are set to zero except t1,1 which is equal to 2K0, the expected coalescence time for a random-mating population of size K0 at mutation-drift equilibrium.
Under the assumption that modern humans colonized the world following routes mainly along landmasses, the populations in the CEPH human genetic–diversity panel most distant from East Africa are in South America, ∼28,000 km away from Ethiopia.7 We thus divided the world into a sequence of stepping-stones consisting of 300 demes, each representing an area of 10,000 km2 (100×100 km). This deme size is similar to that used in previous studies.11 The time needed for the complete process of colonization is largely conditioned by the population-growth rate r. For most of our simulations, there were ∼200–300 generations between the time the colonization of deme 280 (corresponding to the Karitiana in Brazil) and the time of colonization of deme 300 (located at the southern tip of South America). After the last deme was colonized, we let the simulations run for another 200 generations; there is thus a colonization phase followed by a migration phase. The length of this migration phase is based on the assumption that South America was first occupied 10,000–15,000 years ago16 (400–600 generations of 25 years each). It should be noted that the model is not very sensitive to the length of the migration phase. Adding up to 300 generations did not noticeably affect the general fit between actual data and simulated results. The only consequence was an increase in the K and Km estimates (which was always <5%).
This model allows us to obtain expectations for the coalescence times between any pair of genes (the mean age to a common ancestor) for any set of parameter values. Most available evidence suggests microsatellites evolve mainly under a stepwise mutation model (SMM), under which a single repeat is added or deleted by mutation. Under the SMM, the coalescence time Ti within each population i—that is, i∈(1,d)—can be simply estimated from the variance of allele-repeat size within population Xi (see the work of Kimmel and Chakraborty17) as
Using this formula, we estimated coalescence times for all populations from the CEPH panel. We used an effective mutation rate of 7.5×10-4, as advocated elsewhere for this set of loci.5 The individual mutation rate and deviation from the SSM is large for individual loci. However, here, we use a weighted average effective mutation rate over all loci, which has been computed on the same data set.5 This estimate is an effective mutation rate under SSM and thus takes into account deviations from a stepwise model. Since this weighted average is based on a very large number of loci (783), its mean is expected to be highly accurate.
Empirical Data
Individuals from the CEPH panel were split into the same populations as described in the work of Ramachandran et al.5 After elimination of the same duplicates and possibly misclassified individuals, the data set consisted of 1,048 individuals assigned to 54 populations.5 The Surui were removed, since they experienced a severe bottleneck in 1961, when their population size went down to 34 individuals following the spread of diseases brought in by contact with the outside world.18 The two South African Bantu populations are characterized by very small sample sizes. We thus considered the average of the two coalescence times computed separately as a single data point. For the geographical distances, we computed the shortest route through landmasses, also avoiding areas with a mean altitude >2,000 m. We assumed the following land bridges: the route through the Sinai to the Levant as a single connection between Africa and Eurasia, the Bering Strait between Eurasia and the Americas, and a connection between the Malaysian Peninsula and Melanesia. The resulting colonization routes are shown in figure 2. The geographic distances were obtained using an algorithm, based on graph theory, that we developed elsewhere.7 The advantage of this approach over conventional spatial statistics (as used in Geographical Information System software) is that we do not assume the data to be in a Cartesian coordinates system resulting from projection of a spherical surface onto a flat surface. Whereas projections are quite accurate for relatively limited areas, they are problematic for questions that encompass the whole globe.
Figure 2. .

Shortest colonization routes from East Africa (Addis Ababa) along landmasses and high mountain ranges (areas with average elevation >2,000 m) to the populations from the CEPH human genetic–diversity panel. Small blue dots represent populations, the hypothetical origin of modern humans is represented as a large red dot, colonization routes are shown in red, and uncrossable areas with average altitude >2,000 m are brown.
Analyses
There are five parameters in the model that were allowed to vary: time since the spread of modern humans in number of generations t (throughout the article, we assume a generation time of 25 years11,19), growth rate in a newly colonized deme r, migration rate m, carrying capacity of the initial population K0, and carrying capacity of all other demes K. Mutation rate of the microsatellite loci, μ, and the number of demes, d, were considered to be fixed at 7.5×10-4 and 300, respectively. Our simulations were performed in a homogeneous environment, with r, K, and m identical in each deme, regardless of location. Under the assumption that a given genetic data set is the product of a particular evolutionary scenario formed by this set of parameters, one would ideally like to estimate the likelihood of all possible scenarios that can generate the data and choose the one maximizing this likelihood. However, because exploring all possible scenarios is not feasible, given the large number of parameters and the difficulty in computing the likelihood of our model, we restricted our search to a finite number of parameter combinations.
We first considered values of r between 0.2 and 1.2 and of K0 between 250 and 2,000, using a systematic coarse-grid search of the whole parameter space, by incrementing r and K0 by values of 0.1 and 250, respectively. We then performed a fine-grid search for the best-supported parameter value range (0.7⩽r⩽1.0 and 800⩽K0⩽1,200), with increments of 0.05 and 50, respectively. For all combinations of K0 and r, we searched for the values of K and Km (using increments of 5 units) that maximized the fit of the simulation to the actual data, by calculating the sum of square distances between actual data and simulated expectations (corresponding to the sum of squares for the error [SSE]). The parameter set that minimized SSE was considered the best. The goodness of fit was expressed in terms of R2, the proportion of total variance explained by the model. CIs for the parameter values were obtained by considering all models with the Akaike information criteria (AIC) within 4 units from the AIC of the best-fit model, roughly corresponding to the 95% CI, in a least-squares optimization framework,
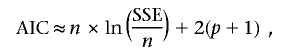 |
where n is the sample size and p is the number of parameters fitted in the model.20 The number of generations needed for the colonization process is a mere consequence of the other variables and is mainly affected by r, which conditions the speed of the wave of advance. We did not consider as plausible any scenario leading to a colonization of the world in <1,500 generations. We carefully evaluated the distribution of residuals by eye. For all simulations comprised within the 95% CI area, residuals were well distributed and thus justified our model fitting by minimizing SSE.
Results
Figure 3 summarizes the fit between analytical expectations and actual data for different sizes of the ancestral founding population in East Africa, K0, and the growth rate of newly colonized demes, r. The contour plot gives the amount of variance in the data explained by the model (R2). The 95% CI is within the red line. The black area represents unrealistic regions of the parameter space, which correspond to a colonization process of the world achieved in <1,500 generations. Averaging all values associated to combinations of K0 and r within the 95% CI yields an initial founding population of 1,064±93 individuals (mean±SD) at mutation-drift equilibrium combined with a growth rate of r=0.86±0.08. The range of parameter space leading to a good fit between simulated and real data includes very high values of r. However, as previously mentioned, extreme values of r also lead to unrealistically short colonization times that can easily be dismissed. For instance, for reasonably large founding population (K0⩾1,200), r>0.9 will invariably lead to a scenario of the colonization of the world in <1,500 generations (37,500 years, under the assumption of generation times of 25 years), which is incompatible with the archaeological evidence.21
Figure 3. .

Contour plot of the fit between simulated and empirical data (R2) for the size of the initial founding population K0 and the growth rate within demes (r). Lighter areas represent better fits between simulations and actual data. The black area represents unrealistic regions of the parameter space, corresponding to a colonization of the world achieved in <1,500 generations. The red line delineates the 95% CI. The best fit between simulations and empirical data (R2>0.72) is obtained for high values of r (∼0.7–1.0) and a founding population, K0, of 800–1,200 individuals.
Our model enables us to infer the optimal values for the carrying capacity of demes (K) and the number of migrants/colonists per deme per generation (Km). Averaging again all values associated with combinations of K0 and r within the 95% CI yields K=751±155 and Km=164±21. Expressed in density, this yields a value of 0.075 effective individuals per km2. Further assuming a 1:3 ratio for effective:census population size,22 we estimate a density of 0.22 individuals per square kilometer. The Km values representing the product of carrying capacity K and the proportion of migrants per population per generation m point to high colonization/migration rates, suggesting that ∼23% of the individuals move from one population to an adjacent one.
By following the same rationale used to estimate the other parameters, in our model, we can evaluate the best-supported time scale for the colonization of the world. Consideration of only the simulations within the 95% CI leads to an average colonization time of the world of 2,243±227 generations. Under the assumption of a 25-year generation interval, this translates into an estimate of the initial expansion of modern humans from East Africa ∼56,063±5,678 years ago. This estimate suggests that humans started expanding shortly before they crossed into Eurasia (an event believed to have occurred ∼45,000–75,000 years ago12,21), a long time after the earliest fossil evidence for anatomically modern humans (∼160,000–195,000 years ago23,24).
So far, we have focused on individual parameters. Equally important, if not more so, is the global fit of the model with the data. In figure 4A, we present expected coalescence times under the best-supported set of parameters against empirical observations, and, in figure 4B, we report gene diversities (heterozygosities). To obtain the expectation for gene diversities that are commonly used in population genetics, we transformed the mutation rate under SMM to an infinite-allele model (IAM) equivalent. The IAM equivalent mutation rate (μIAM) here is 2×10-4, which is obtained by assuming an effective size for the entire human population of 10,000 effective individuals. The general fit for both coalescence times and gene diversities is remarkable. It is also noteworthy that the expectations for coalescence times and gene diversities do not decrease linearly with geographic distance. This is due to populations in the middle of the sequence of stepping-stones having higher effective neighborhoods (i.e., they receive more migrants). An important corollary stemming from this nonlinearity is that use of linear regressions on gene diversities5 might not allow correct inference of the geographic origin of modern humans.
Figure 4. .

Illustration of coalescence times (A) and gene diversities (B) plotted against geographic distance from East Africa, computed through landmasses. The continuous lines represent analytical expectations from the model, and the unblackened circles represent actual values for 52 populations from the CEPH human genetic–diversity panel. In this figure, parameter values for the simulations are as follows: K0=1,000, r=0.85, K=800, Km=150, and the number of generations=2,392.
Discussion
Over recent years, a near consensus has emerged in favor of a recent single origin of modern humans in East Africa.9–13 However, considerable uncertainty clouds any more detailed aspect of human-colonization history. Here, we estimated key parameters of the spread of modern humans, using a model of isolation by distance, which is supported by the observation that genetic diversity decreases smoothly with geographic distance along landmasses from East Africa.7,8 Our general model fits remarkably well with the general pattern of empirical data based on 52 populations from the CEPH human genetic–diversity panel genotyped at 783 autosomal microsatellites (fig. 4). Our results point to an expansion of modern humans ∼56,000 years ago, from a founding population of 1,000 effective individuals. We further obtained very high population-growth rates within newly colonized demes.
We estimate an ancestral founding population of 1,064±93 effective individuals. Under the assumption of a 1:3 ratio for effective:census population size,22 this would suggest an ancestral population of ∼3,000 individuals. Although this value may seem small, it is in line with some of the previous extremely low estimates that were based on autosomal and Y-chromosome microsatellite loci.25,26 It is likely that other human populations lived in Africa at the same time but did not contribute to the colonization of the world.25,27 Our results thus suggest that the demographic expansion of anatomically modern humans started from a limited geographic area. Such a scenario is compatible with the patterns observed in mtDNA and Y chromosome, where the diversity found outside Africa represents only a fraction of the diversity seen among African haplotypes.21,28
Our results point to very fast growth in newly colonized demes. Our best-supported rates of increase (∼0.86±0.08 in a logistic growth model) are at the higher end of available values estimated for human hunter-gatherer populations (0.3<r<0.9).29–31 It is, however, important to realize that our r values affect growth rate only in newly colonized environments. It is likely that the early settlers benefited from extremely favorable conditions, with an essentially unlimited supply of naive prey, as suggested by the catastrophic faunal extinctions that have occurred in the wake of human arrival in previously uninhabited regions of the world.32
Our model predicts a density of 0.22 individuals per square kilometer, well within the 0.01–0.35 range estimated for ancient and modern hunter-gatherer societies.31,33,34 The Km values representing the product of carrying capacity K and the proportion of migrants per population per generation indicate high migration rates, suggesting that ∼23% of the individuals moved from one population to another. This figure should be evaluated with circumspection, since its biological interpretation is not straightforward. There are three features in our model that may make our parameter m an overestimate of the real migration rate. First, we did not allow for long-distance migration, which would be far more effective at homogenizing demes. Second, we allowed for migration from a deme only when it had reached its carrying capacity. Third, we did not separate migration (the exchange of individuals between demes at carrying capacity) and colonization (movement of individuals to nonsaturated adjacent demes).
The wave of migration out of Africa is suggested to have occurred <100,000 years ago and to have led to the subsequent colonization of the entire world, with the replacement of previously established human species, such as Neanderthals in Europe.12,35,36 Archaeological findings provide potential dates for the key events. The oldest remains of modern humans, which presumably pinpoint the origin of our species, have been found in eastern and southern Ethiopia and have been dated at 160,000 and 195,000 years, respectively.23,24 Although there have been several attempts to quantify the size of the population that moved out of Africa, fewer attempts have been made to estimate the starting date of the colonization process. Zhivotovsky and colleagues25 give an estimate of 71–142 thousand years ago (ky). Our evaluation is much lower (56,063±5,678 years).
Zhivotovsky et al.25 also provide time estimates for the expansion in population size for African farmer or pastoralist populations (35.3 ky), Eurasian (25.3 ky), and East Asian populations (17.6 ky). We can contrast their estimates with our model estimates for when these populations started expanding. To do so, we can evaluate when these three areas are colonized in our model. Following this rationale, we obtained estimates for the expansion of ∼48 ky for African farmers, ∼40 ky for Eurasians, and ∼36 ky for East Asians. The two series of estimates are highly divergent. This is not entirely surprising, since the times of expansion do not have the same meaning in the two models. In our model, population expansion starts as soon as the area is colonized and lasts until carrying capacity is reached. In the model by Zhivotovsky et al.,25 expansion is decoupled from colonization and is meant to capture more-recent hypothetical events of population growth. For instance, their estimate of expansion of African hunter-gatherers is very recent (4.3 ky), whereas we obtain a figure similar to that obtained for African farmers (∼50 ky). This large difference in the time of population expansion between African farmers and hunter-gatherers in the work of Zhivotovsky et al. is intriguing and cannot be explained by a demographic effect of farming alone, since the estimated demographic expansion of African farmers predates by 25,000 years the first evidence of agriculture.
A calibration point that has attracted much interest is the time of exit from Africa. The first evidence of modern humans outside Africa comes from Israel and has been dated at 80,000–100,000 years ago.37 However, this observation of modern humans in the Middle East is isolated and may represent an early offshoot that died out. The later, successful migration(s) out of Africa are believed to have occurred 45,000–75,000 years ago.12,21,28 Our results support this view, since the out-of-Africa event corresponding to populations moving through the Sinai to the Levant in our model is predicted to have happened 45,000–55,000 years ago. When we computed our shortest distance through landmasses for the various populations (fig. 2), we did not consider a possible southern route through the Horn of Africa, along the tropical coast of the Indian Ocean to Southeast Asia and Australasia.21,38 However, both routes give similar geographic distances from East Africa to the 52 populations analyzed, and the results are essentially unaffected. The same is true for the colonization of Melanesia, where we considered a route through the Malaysian Peninsula rather than the Indian subcontinent.
Beyond single-parameters estimates, our model is remarkably effective at fitting empirical data (fig. 4). This may come as a surprise to those who assume that human-settlement history was so complex that it cannot be captured by simple models.39 Our model is indeed simple, since it considers only within-population coalescence times. We further neglected key events such as spatial and temporal environmental variation. Our results thus suggest that various environmental factors tend to be spatially relatively homogeneous for human migration patterns, when considered over a large geographic distance. We fully acknowledge that any tractable population-genetics model will come at the cost of some idiosyncrasies. However, the strength of our approach stems from the coupling of a formal coalescence-times model with a sophisticated geographically explicit treatment of migration routes.7 It seems that this mixture of a population-genetics model with dynamic demography superimposed on explicit geography creates a surprisingly powerful tool. Earlier related work focused on specific questions by fixing other parameters.11,36 Our framework allows all parameters to evolve freely; thus, we argue that it might be the precursor of a generation of tools that will greatly help us to understand the details of colonization history of humans as well as other species.
Acknowledgments
The work was supported by the Biotechnology and Biological Sciences Research Council. H.L. acknowledges support from the Cambridge Overseas Trust.
References
- 1.Rosenberg NA, Pritchard JK, Weber JL, Cann HM, Kidd KK, Zhivotovsky LA, Feldman MW (2002) Genetic structure of human populations. Science 298:2381–2385 10.1126/science.1078311 [DOI] [PubMed] [Google Scholar]
- 2.Cann HM, de Toma C, Cazes L, Legrand MF, Morel V, Piouffre L, Bodmer J, et al (2002) A human genome diversity cell line panel. Science 296:261–262 10.1126/science.296.5566.261b [DOI] [PubMed] [Google Scholar]
- 3.Relethford JH (2004) Global patterns of isolation by distance based on genetic and morphological data. Hum Biol 76:499–513 10.1353/hub.2004.0060 [DOI] [PubMed] [Google Scholar]
- 4.Manica A, Prugnolle F, Balloux F (2005) Geography is a better determinant of human genetic differentiation than ethnicity. Hum Genet 118:366–371 10.1007/s00439-005-0039-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramachandran S, Deshpande O, Roseman CC, Rosenberg NA, Feldman MW, Cavalli-Sforza LL (2005) Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci USA 102:15942–15947 10.1073/pnas.0507611102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serre D, Pääbo S (2004) Evidence for gradients of human genetic diversity within and among continents. Genome Res 14:1679–1685 10.1101/gr.2529604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prugnolle F, Manica A, Balloux F (2005) Geography predicts neutral genetic diversity of human populations. Curr Biol 15:R159–R160 10.1016/j.cub.2005.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prugnolle F, Manica A, Charpentier M, Guégan J, Guernier V, Balloux F (2005) Pathogen-driven selection and worldwide HLA class I diversity. Curr Biol 15:1022–1027 10.1016/j.cub.2005.04.050 [DOI] [PubMed] [Google Scholar]
- 9.Harpending H, Rogers AR (2000) Genetic perspectives on human origins and differentiation. Annu Rev Genomics Hum Genet 1:361–385 10.1146/annurev.genom.1.1.361 [DOI] [PubMed] [Google Scholar]
- 10.Cavalli-Sforza LL, Feldman MW (2003) The application of molecular genetic approaches to the study of human evolution. Nat Genet 33:266–275 10.1038/ng1113 [DOI] [PubMed] [Google Scholar]
- 11.Ray N, Currat M, Berthier P, Excoffier L (2005) Recovering the geographic origin of early modern humans by realistic and spatially explicit simulations. Genome Res 15:1161–1167 10.1101/gr.3708505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stringer C, Andrews P (1988) Genetic and fossil evidence for the origin of modern humans. Science 239:1263–1268 [DOI] [PubMed] [Google Scholar]
- 13.Stringer C (2002) Modern human origins: progress and prospects. Philos Trans R Soc Lond B Biol Sci 357:563–579 10.1098/rstb.2001.1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Austerlitz F, Jung-Muller B, Godelle B, Gouyon P (1997) Evolution of coalescence times, genetic diversity and structure during colonization. Theor Popul Biol 51:148–164 10.1006/tpbi.1997.1302 [DOI] [Google Scholar]
- 15.Excoffier L, Schneider S (1999) Why hunter-gatherer populations do not show signs of Pleistocene demographic expansions. Proc Natl Acad Sci USA 96:10597–10602 10.1073/pnas.96.19.10597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dillehay TD (1999) The late Pleistocene cultures of South America. Evol Anthropol 7:206–216 [DOI] [Google Scholar]
- 17.Kimmel M, Chakraborty R (1996) Measures of variation at DNA repeat loci under a general stepwise mutation model. Theor Popul Biol 50:345–367 10.1006/tpbi.1996.0035 [DOI] [PubMed] [Google Scholar]
- 18.Laraia RB, Da Matta R (1979) Índios e castanheiros: a empresa extrativista e os índios no Médio Tocantins. Paz e Terra, Rio de Janeiro, p 208 [Google Scholar]
- 19.Tremblay M, Vézina H (2000) New estimates of intergenerational time intervals for the calculation of age and origins of mutations. Am J Hum Genet 66:651–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burnham KP, Anderson DR (1998) Model selection and inferences. Springer Verlag, New York [Google Scholar]
- 21.Macaulay V, Hill C, Achilli A, Rengo C, Clarke D, Meehan W, Blackburn J, Semino O, Scozzari R, Cruciani F, Taha A, Shaari NK, Raja J, Ismail P, Zainuddin Z, Goodwin W, Bulbeck D, Bandelt HJ, Oppenheimer S, Torroni A, Richards M (2005) Single, rapid coastal settlement of Asia revealed by analysis of complete mitochondrial genomes. Science 308:1034–1036 10.1126/science.1109792 [DOI] [PubMed] [Google Scholar]
- 22.Cavalli-Sforza LL, Menozzi P, Piazza A (1994) The history and geography of human genes. Princeton University Press, Princeton, NJ [Google Scholar]
- 23.White TD, Asfaw B, DeGusta D, Gilbert H, Richards GD, Suwa G, Howell FC (2003) Pleistocene Homo sapiens from Middle Awash, Ethiopia. Nature 423:742–747 10.1038/nature01669 [DOI] [PubMed] [Google Scholar]
- 24.McDougall I, Brown FH, Fleagle JG (2005) Stratigraphic placement and age of modern humans from Kibish, Ethiopia. Nature 433:733–736 10.1038/nature03258 [DOI] [PubMed] [Google Scholar]
- 25.Zhivotovsky LA, Rosenberg NA, Feldman MW (2003) Features of evolution and expansion of modern humans, inferred from genomewide microsatellite markers. Am J Hum Genet 72:1171–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pritchard JK, Seielstad MT, Perez-Lezaun A, Feldman MW (1999) Population growth of human Y chromosomes: a study of Y chromosome microsatellites. Mol Biol Evol 16:1791–1798 [DOI] [PubMed] [Google Scholar]
- 27.Satta Y, Takahata N (2004) The distribution of the ancestral haplotype in finite stepping-stone models with population expansion. Mol Ecol 13:877–886 10.1046/j.1365-294X.2003.02069.x [DOI] [PubMed] [Google Scholar]
- 28.Underhill PA, Shen PD, Lin AA, Jin L, Passarino G, Yang W, Kauffman E, Bonne-Tamir B, Bertranpetit J, Francalacci P, Ibrahim M, Jenkins T, Kidd JR, Mehdi SQ, Seielstad MT, Wells RS, Piazza A, Davis RW, Feldman MW, Cavalli-Sforza LL, Oefner PJ (2000) Y chromosome sequence variation and the history of human populations. Nat Genet 26:358–361 10.1038/81685 [DOI] [PubMed] [Google Scholar]
- 29.Alroy J (2001) A multispecies overkill simulation of the end-Pleistocene megafaunal mass extinction. Science 292:1893–1896 10.1126/science.1059342 [DOI] [PubMed] [Google Scholar]
- 30.Pennington R (2001) Hunter-gatherer demography. In: Panter-Brick C, Layton R (eds) Hunter-gatherers: an interdisciplinary perspective. Cambridge University Press, Cambridge, United Kingdom, pp 170–204 [Google Scholar]
- 31.Steele J, Adams JM, Sluckin T (1998) Modeling Paleoindian dispersals. World Archaeol 30:286–305 [Google Scholar]
- 32.Burney DA, Flannery TF (2005) Fifty millennia of catastrophic extinctions after human contact. Trends Ecol Evol 20:395–401 10.1016/j.tree.2005.04.022 [DOI] [PubMed] [Google Scholar]
- 33.Bocquet-Appel JP, Demars PY (2000) Neanderthal contraction and modern human colonization of Europe. Antiquity 74:544–552 [Google Scholar]
- 34.Binford LR (2001) Constructing frames of reference: an analytical method for archaeological theory building using ethnographic and environmental data sets. University of California Press, Berkeley [Google Scholar]
- 35.Cann RL, Stoneking M, Wilson AC (1987) Mitochondrial DNA and human evolution. Nature 325:31–36 10.1038/325031a0 [DOI] [PubMed] [Google Scholar]
- 36.Currat M, Excoffier L (2004) Modern humans did not admix with Neanderthals during their range expansion into Europe. PLoS Biol 2:2264–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDermott F, Grun R, Stringer CB, Hawkesworth CJ (1993) Mass-spectrometric U-series dates for Israeli Neanderthal early-modern hominid sites. Nature 363:252–255 10.1038/363252a0 [DOI] [PubMed] [Google Scholar]
- 38.Lahr MM, Foley RA (1998) Multiple dispersals and modern human origins. Evol Anthropol 3:48–60 10.1002/evan.1360030206 [DOI] [Google Scholar]
- 39.Foley RA (1998) The context of human genetic evolution. Genome Res 8:339–347 [DOI] [PubMed] [Google Scholar]