

Abstract
Background
Multiple sclerosis (MS) often leads to severe neurological disability and a serious decline in quality of life. The ideal target of disease‐modifying therapy for MS is to prevent disability worsening and improve quality of life. Dimethyl fumarate is considered to have an immunomodulatory activity and neuroprotective effect. It has been approved by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency as a first‐line therapy for adult patients with relapsing‐remitting MS (RMSS).
Objectives
To assess the benefit and safety of dimethyl fumarate as monotherapy or combination therapy versus placebo or other approved disease‐modifying drugs (interferon beta, glatiramer acetate, natalizumab, mitoxantrone, fingolimod, teriflunomide, alemtuzumab) for patients with MS.
Search methods
The Trials Search Co‐ordinator searched the Trials Specialised Register of the Cochrane Multiple Sclerosis and Rare Diseases of the Central Nervous System Group (4 June 2014). We checked reference lists of published reviews and retrieved articles and searched reports (2004 to June 2014) from the MS societies in Europe and America. We also communicated with investigators participating in trials of dimethyl fumarate and the Biogen Idec Medical Information.
Selection criteria
We included randomised, controlled, parallel‐group clinical trials (RCTs) with a length of follow‐up equal to or greater than one year evaluating dimethyl fumarate, as monotherapy or combination therapy, versus placebo or other approved disease‐modifying drugs for patients with MS without restrictions regarding dosage, administration frequency and duration of treatment.
Data collection and analysis
We used the standard methodological procedures of The Cochrane Collaboration. Two review authors independently assessed trial quality and extracted data. Disagreements were discussed and resolved by consensus among the review authors. We contacted the principal investigators of included studies for additional data or confirmation of data.
Main results
Two RCTs were included, involving 2667 adult patients with RRMS to evaluate the efficacy and safety of two dosages of dimethyl fumarate (240 mg orally three times daily or twice daily) by direct comparison with placebo for two years. Among them, a subsample of 1221 (45.8%) patients were selected to participate in MRI evaluations by each study site with MRI capabilities itself. No powered head‐to‐head study with an active treatment comparator has been found. Meta‐analyses showed that dimethyl fumarate both three times daily and twice daily reduced the number of patients with a relapse (risk ratio (RR) 0.57, 95% confidence interval (CI) 0.50 to 0.66, P < 0.00001 and 0.64, 95% CI 0.54 to 0.77, P < 0.00001, respectively) or disability worsening (RR 0.70, 95% CI 0.57 to 0.87, P = 0.0009 and 0.65, 95% CI 0.53 to 0.81, P = 0.0001, respectively) over two years, compared to placebo. The treatment effects were decreased in the likely‐case scenario analyses taking the effect of dropouts into consideration. Both dosages also reduced the annualised relapse rate. Data of active lesions on MRI scans were not combined because there was a high risk of selection bias for MRI outcomes and imprecision of MRI data in both studies, as well as an obvious heterogeneity between the studies. In terms of safety profile, both dosages increased the risk for adverse events and the risk for drug discontinuation due to adverse events. The most common adverse events included flushing and gastrointestinal events (upper abdominal pain, nausea and diarrhoea). Uncommon adverse events included lymphopenia and leukopenia, but they were more likely to happen with dimethyl fumarate than with placebo (high dosage: RR 5.25, 95% CI 2.20 to 12.51, P = 0.0002 and 5.23, 95% CI 2.47 to 11.07, P < 0.0001, respectively; low dosage: RR 5.69, 95% CI 2.40 to 13.46, P < 0.0001 and 6.53, 95% CI 3.13 to 13.64, P < 0.00001, respectively). Both studies had a high attrition bias resulting from the unbalanced reasons for dropouts among groups. Quality of evidence for relapse outcome was moderate, but for disability worsening was low.
Authors' conclusions
There is moderate‐quality evidence to support that dimethyl fumarate at a dose of 240 mg orally three times daily or twice daily reduces both the number of patients with a relapse and the annualised relapse rate over two years of treatment in comparison with placebo. However, the quality of the evidence to support the benefit in reducing the number of patients with disability worsening is low. There is no high‐quality data available to evaluate the benefit on MRI outcomes. The common adverse effects such as flushing and gastrointestinal events are mild‐to‐moderate for most patients. Lymphopenia and leukopenia are uncommon adverse events but significantly associated with dimethyl fumarate. Both dosages of dimethyl fumarate have similar benefit and safety profile, which supports the option of low‐dose administration. New studies of high quality and long‐term follow‐up are needed to evaluate the benefit of dimethyl fumarate on prevention of disability worsening and to observe the long‐term adverse effects including progressive multifocal leukoencephalopathy.
Plain language summary
Dimethyl fumarate (BG‐12) first line oral treatment for people with multiple sclerosis
Background
Dimethyl fumarate was approved by the U.S. Food and Drug Administration (FDA) and the European Medicines Agency for adults with relapsing‐remitting multiple sclerosis (RRMS). It does not require that any other medication be tried before dimethyl fumarate is prescribed. Although data from non Cochrane reviews are available, it is important to systematically evaluate its efficacy and safety as monotherapy versus placebo.
Study characteristics
We searched medical databases for studies in which participants were randomly assigned to dimethyl fumarate or a control drug (randomised controlled trials). The efficacy of this therapy was considered in terms of occurrence of relapses and progression of the disease.
Key results and quality of evidence
We found moderate quality evidence that both dosages of dimethyl fumarate reduce the number of people with RRMS having relapse after two years of treatment, while there is low quality evidence showing that the medicine reduces the number of people who experience worsening disability at the end of two years. Common adverse effects such as flushing and gastrointestinal events (diarrhoea, nausea and upper abdominal pain) are mild to moderate for most of patients. Dimethyl fumarate can have an effect on the body's immune system by causing a drop in the number of the white blood cells which help to fight infection. More people in the groups treated with dimethyl fumarate experienced this than they did with placebo. We found moderate quality that people were more likely to leave the study early because of adverse events if they were treated with dimethyl fumarate than with placebo.
Summary of findings
Summary of findings for the main comparison. Dimethyl fumarate for multiple sclerosis.
| Dimethyl fumarate for multiple sclerosis | ||||||
| Patient or population: Patients with relapsing‐remitting multiple sclerosis Settings: United States, Germany, Poland, India, Canada, France, etc Intervention: Dimethyl fumarate at a dose of 240 mg orally twice daily | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Dimethyl fumarate | |||||
| The proportion of patients with at least one relapse at two years Follow‐up: 2 years | Study population | RR 0.64 (0.54 to 0.77) | 1540 (2 studies) | ⊕⊕⊕⊝ moderate1 | ||
| 437 per 1000 | 280 per 1000 (236 to 337) | |||||
| The proportion of patients with disability worsening at two years Follow‐up: 2 years | Study population | RR 0.65 (0.53 to 0.81) | 1539 (2 studies) | ⊕⊕⊝⊝ low1,2 | ||
| 223 per 1000 | 145 per 1000 (118 to 181) | |||||
| The proportion of patients who discontinued study drug because of adverse events excluding relapses at two years Follow‐up: 2 years | Study population | RR 2.18 (1.56 to 3.06) | 1540 (2 studies) | ⊕⊕⊕⊝ moderate1 | ||
| 58 per 1000 | 127 per 1000 (91 to 179) | |||||
|
The proportion of patients with lymphopenia at two years Follow‐up: 2 years |
Study population | RR 5.69 (2.40 to 13.46) | 1540 (2 studies) | ⊕⊕⊕⊝ moderate1 | ||
| 8 per 1000 | 44 per 1000 (19 to 105) | |||||
| *The basis for the assumed risk (e.g. the median placebo group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio. | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 A high rate of dropouts existed and reasons of dropouts were unbalanced between arms 2 Disability worsening was confirmed at 3 months of follow‐up
Background
Description of the condition
Multiple sclerosis (MS) is a chronic immune‐mediated disease of the central nervous system. It is pathologically characterised by inflammation, demyelination, and axonal and neuronal loss. Clinically it is characterised by recurrent relapses or progression, or both, typically striking adults during the primary productive time of their lives and ultimately leading to severe neurological disability.
There are four clinical phenotypes of MS. Initially, more than 80% of individuals with MS experience a relapsing‐remitting disease course (RRMS), characterised by clinical exacerbations of neurological symptoms followed by complete or incomplete remission (Lublin 1996). After 10 to 20 years, or a median age of 39 years, about half of these people gradually accumulate irreversible neurological deficits, with or without clinical relapses (Confavreux 2006), which is known as secondary progressive MS (SPMS). Another 10% to 20% of individuals with MS are diagnosed with primary progressive MS (PPMS), clinically defined as a disease course without any clinical attacks or remission from onset (Lublin 1996). A significantly rarer form is progressive relapsing MS (PRMS), which initially presents as PPMS; however, during the course of the disease these individuals develop true neurological exacerbations (Tullman 2004).
MS causes a major socioeconomic burden, both for the individual patient and for society. Increasing costs and decreasing quality of life are associated with advancing disease severity, disease progression and relapses (Karampampa 2012a; Karampampa 2013b). From a patient's perspective, a MS relapse is associated with a significant increase in economic costs as well as a decline in health‐related quality of life (HRQoF) and functional ability (Oleen‐Burkey 2012). Effective treatment that reduces relapse frequency and prevents progression could have an impact both on costs and HRQoF, and may help to reduce the social burden of MS (Karampampa 2012c).
Natalizumab and interferon (IFN) β‐1a (IFNβ‐1a) (Rebif™) have been shown to be superior to mitoxantrone, glatiramer acetate, IFNβ‐1b (Betaseron™) and IFNβ‐1a (Avonex™) for preventing clinical relapses in RRMS in the short term (24 months) compared to placebo, based on high‐quality evidence. However, they are associated with long‐term serious adverse events (SAEs) and their benefit‐risk balance might be unfavourable (Filippini 2013). Furthermore, administration of IFNβ is not associated with a reduction in the worsening of disability among patients with RRMS (Shirani 2012). Therefore, there is a need for safer and more effective drugs with new modes of action that lead to anti‐inflammation and neuroprotection in MS.
Inflammation and oxidative stress are thought to cause tissue damage in MS. Therefore, oxidative stress and antioxidative pathways are important in MS pathophysiology (Gilgun‐Sherki 2004; Lee 2012), and novel therapeutics that enhance cellular resistance to free radicals could prove useful for MS treatment. Recent data support this important role of antioxidative pathways for tissue protection in progressive MS, particularly by activation of the transcription factor nuclear (erythroid‐derived 2)‐related factor 2 (Nrf2) antioxidant pathway (Johnson 2010), which is not yet targeted by other disease‐modifying therapies for MS (Linker 2011).
Description of the intervention
An oral formulation of dimethyl fumarate (BG‐12) has shown promising results for RRMS in clinical trials, combining anti‐inflammatory and possibly clinically relevant neuroprotective effects with the convenience of oral administration. An exploratory, prospective, open‐label pilot study showed that fumaric acid esters produced significant reductions from baseline in the number and volume of gadolinium‐enhancing lesions in patients with RRMS (Schimrigk 2006). A phase II study with oral BG‐12 revealed a dose‐dependent, significant reduction in brain lesion activity. Oral BG‐12 at a dose of 240 mg three times daily significantly reduced the number of new gadolinium‐enhancing lesions, new or enlarging T2‐hyperintense and new T1‐hypointense lesions, and the annualised relapse rate (ARR) in patients with RRMS (Kappos 2008). A randomised, double‐blind, placebo‐controlled phase III study (DEFINE) showed that both oral BG‐12 dosages (at a dose of 240 mg twice daily and 240 mg three times daily) significantly reduced the proportion of patients who had a relapse, the ARR, the rate of disability worsening and the number of lesions in patients with RRMS (Gold 2012) when compared with placebo. Another phase III study (CONFIRM) showed that both oral BG‐12 dosages and glatiramer acetate (subcutaneous daily injections of 20 mg) significantly reduced the ARR and the numbers of new or enlarging T2‐weighted hyperintense lesions and new T1‐weighted hypointense lesions in patients with RRMS, compared with placebo, but the reductions in disability worsening with twice‐daily BG‐12, thrice‐daily BG‐12 and glatiramer acetate were not significant (Fox 2012). Post hoc comparisons of BG‐12 with glatiramer acetate showed significant differences in the ARR (thrice‐daily BG‐12), new or enlarging T2‐weighted hyperintense lesions (both BG‐12 dosages) and new T1‐weighted hypointense lesions (thrice‐daily BG‐12). Furthermore, BG‐12 had positive effects on health‐related quality of life in patients with RRMS.
Dimethyl fumarate (Tecfidera™) is a new oral drug approved by the U.S. Food and Drug Administration (FDA) in 2013 as a first‐line therapy for adult patients with relapsing MS, and in the same year, it was also approved in Canada and in Australia. In 2014, it was approved by the European Medicines Agency (EMA) as a first‐line oral treatment for adult patients with RRMS. The proper usage indicates dimethyl fumarate capsules are taken orally twice per day, with a one‐week reduced starting dose (120 mg twice a day), and thereafter a maintenance dose (240 mg twice a day) (National MS Society 2013).
How the intervention might work
Several lines of research have demonstrated immunomodulatory but also neuroprotective effects of fumaric acid esters, as shown in vitro as well as in experimental models of MS (Lukashev 2007). Fumaric acid esters include methyl hydrogen fumarate and dimethyl fumarate. Immunomodulatory concentrations of dimethyl fumarate can reduce oxidative stress without altering neuronal network activity (Albrecht 2012). In the acute phase of experimental autoimmune encephalomyelitis, treatment with dimethyl fumarate resulted in a significant reduction of macrophage/microglia infiltration in inflamed lesions (Schilling 2006). In an in vitro model of brain inflammation, dimethyl fumarate inhibited microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, interleukin (IL)‐1β, tumour necrosis factor (TNF)‐α and IL‐6 (Wilms 2010). Dimethyl fumarate and its primary metabolite, monomethyl fumarate, are cytoprotective of neurons and astrocytes against oxidative stress‐induced cellular injury and loss, potentially by induction of the transcription factor Nrf‐2 and up‐regulation of an Nrf2‐dependent antioxidant response (Scannevin 2012). In addition, dimethyl fumarate treatment for type 1 helper T cells (Th1) and T helper type 17 (Th17)‐mediated MS induces IL‐4‐producing Th2 cells in vivo and generates type II dendritic cells that produce IL‐10 instead of IL‐12 and IL‐23 (Ghoreschi 2011). Dimethyl fumarate inhibits maturation of dendritic cells and subsequently Th1 and Th17 cell differentiation by suppression of both nuclear factor κB (NF‐κB) and extracellular signal‐regulated kinase 1 and 2 (ERK1/2) and mitogen stress‐activated kinase 1 (MSK1) (ERK1/2‐MSK1) signalling (Peng 2012).
Dimethyl fumarate, the active component of BG00012 (BG‐12), is absorbed almost exclusively in the small intestine within two hours after oral administration and is rapidly hydrolysed by esterases to its metabolite, monomethyl fumarate, in the intestinal mucosa (Litjens 2004). Dimethyl fumarate possesses a pharmacological half‐life of about 12 minutes and does not show any binding activity to serum proteins, which may further contribute to its rapid turnover in the circulation (Lee 2008). There is no evidence for a cytochrome P450‐dependent metabolism of fumaric acid esters in the liver (Lee 2008).
Why it is important to do this review
A recent non‐Cochrane review based on indirect comparison has shown that dimethyl fumarate offers an effective oral treatment option for patients with RRMS, with an overall promising efficacy and safety profile, compared to IFNβ‐1a, IFNβ‐1b, glatiramer acetate, fingolimod, natalizumab and teriflunomide (Hutchinson 2014). Another non‐Cochrane review evaluated the efficacy and safety of dimethyl fumarate for RRMS based on direct comparison with placebo or glatiramer acetate, with results showing that dimethyl fumarate (at both dosages) was more effective in reducing relapse, disability worsening and gadolinium‐enhancing lesions at two years (Kawalec 2014). However, the authors did not fully consider the potential impact of bias on the internal validity of the results and did not conduct a comprehensive analysis of the quality of evidence.
Objectives
To assess the absolute and comparative efficacy and safety of dimethyl fumarate as monotherapy or combination therapy versus placebo or other approved disease‐modifying drugs (IFN‐β, glatiramer acetate, natalizumab, mitoxantrone, fingolimod, teriflunomide, alemtuzumab) for patients with MS.
Methods
Criteria for considering studies for this review
Types of studies
All randomised, controlled, parallel‐group clinical trials (RCTs) evaluating dimethyl fumarate, as monotherapy or combination therapy, versus placebo or other approved disease‐modifying drugs for patients with MS. We excluded trials with follow‐up of less than one year.
Types of participants
We included patients aged 18 to 60 years with a definite diagnosis of MS as defined according to Poser's (Poser 1983) or McDonald's (McDonald 2001; Polman 2005; Polman 2011) criteria, any clinical phenotypes categorised according to the classification of Lublin and Reingold (Lublin 1996), and an Expanded Disability Status Scale (EDSS) (Kurtzke 1983) score of 6.0 or lower.
Types of interventions
Experimental intervention
Treatment with dimethyl fumarate orally, as monotherapy or combination therapy, without restrictions regarding dosage, administration frequency or duration of treatment.
Control intervention
Placebo or an approved disease‐modifying drug.
Types of outcome measures
Primary outcomes
We assessed the following primary outcomes.
Benefit
The proportion of patients with at least one relapse at one year or two years. Confirmed relapse was defined as the occurrence of new or worsening of previously stable symptoms, not associated with fever or infection, occurring at least 30 days after the onset of a preceding relapse, lasting longer than 24 hours and that were accompanied by new objective neurological findings according to a neurologist's evaluation.
The proportion of patients with disability worsening as assessed by the EDSS at one year or two years. Disability worsening was defined as an increase in the EDSS score of at least 1.0 point in patients with a baseline score of 1.0 or higher or an increase of at least 1.5 points in patients with a baseline score of 0, with the increased score sustained for six months. We used the data where disability worsening was confirmed in less than six months, however we downgraded the study for indirectness of evidence when we performed the GRADE assessment.
Safety
The proportion of patients with at least one adverse event (AE), the proportion of patients with at least one SAE and the proportion of patients who discontinued the study drug because of AEs at one year and two years.
Secondary outcomes
We assessed the following secondary outcomes.
The ARR at one year or two years, defined as the mean number of confirmed relapses per patient, adjusted for the duration of follow‐up to annualise it.
The number (rate) of gadolinium‐enhancing T1‐weighted lesions at one year or two years.
The number (rate) of new or enlarging T2‐weighted hyperintense lesions at one year or two years.
The percentage brain volume change at one year or two years.
Mean change in HRQoL from baseline to one year or two years. We accepted the following scales: the Medical Outcomes Study (MOS) 36‐Item Short‐Form Health Survey (SF‐36) (Ware 1992), the Multiple Sclerosis Quality of Life‐54 (MSQOL‐54) (Vickrey 1995), the Multiple Sclerosis Quality of Life Inventory (MSQLI) (Fischer 1999) or the Functional Assessment of Multiple Sclerosis (FAMS) (Cella 1996).
Search methods for identification of studies
A systematic search with no restrictions was conducted to identify all relevant published and unpublished RCTs. We applied no language restrictions to the search.
Electronic searches
The Trials Search Co‐ordinator searched the Trials Specialised Register of the Cochrane Multiple Sclerosis and Rare Diseases of the Central Nervous System Group (4 June 2014) which contains the following:
The Cochrane Central Register of Controlled Trials (CENTRAL) (2014, Issue 6)
MEDLINE (PubMed) (1966 to 4 June 2014)
EMBASE (EMBASE.com) (1974 to 4 June 2014)
Cumulative Index to Nursing and Allied Health Literature (CINAHL) (EBSCO host) (1981 to 4 June 2014)
Latin American and Caribbean Health Science Information Database (LILACS) (Bireme) (1982 to 4 June 2014)
Clinical trials registries (http://clinicaltrials.gov/)
World Health Organization (WHO) International Clinical Trials Registry Portal (apps.who.int/trialsearch/)
Information on the Trial Register of the Review Group and details of search strategies used to identify trials can be found in the 'Specialised Register' section within the Cochrane Multiple Sclerosis and Rare Diseases of the Central Nervous System Group's module.
The keywords used to search for trials for this review are listed in Appendix 1.
Searching other resources
We checked reference lists of published reviews and retrieved articles for additional trials. We searched reports (2004 to June 2014) from the MS Societies (National Multiple Sclerosis Society (United States, United Kingdom)) (http://www.nationalmssociety.org/) and the Congress of the European and Americas Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS (http://www.ectrims.eu/) and ACTRIMS (http://www.actrims.org/)). We communicated personally with investigators participating in trials of dimethyl fumarate. We also contacted the biotechnology company Biogen Idec in an effort to identify further studies (http://www.biogenidec.com).
Data collection and analysis
Selection of studies
Two review authors (Xu and He) independently screened titles and abstracts of the citations retrieved by the literature search for inclusion or exclusion. We obtained the available full texts of potentially relevant studies for further assessment. We independently evaluated the eligibility of these studies (on the basis of information available in the published data) and listed papers that did not meet the inclusion criteria in the 'Characteristics of excluded studies' table with the reasons for exclusion. We resolved any disagreement regarding inclusion by discussion or by referral to a third review author (Dong) if necessary.
Data extraction and management
Two review authors (Xu, He) independently extracted information and data from the selected trials using standardised forms, including information about eligibility criteria, methods (study design, total study duration, sequence generation, allocation concealment, blinding and other concerns about bias), participants (total number, setting, diagnostic criteria, age, sex and country), interventions (total number of intervention groups and specific intervention) and outcomes (outcomes and time points, outcome definition and unit of measurement), results (number of participants allocated to each intervention group, sample size, missing participants and summary data for each intervention group), and funding source. We contacted the principal investigators of included studies and the Biogen Idec Medical Information in order to request additional data or confirmation of methodological aspects of the study. We discussed and resolved disagreements by consensus among the review authors.
Assessment of risk of bias in included studies
We based the methodological criteria on theCochrane Handbook for Systematic Review of Interventions Version 5.1.0 (Higgins 2011). Two review authors (Xu and He) independently evaluated the methodological quality of the studies using the 'Risk of bias' tool Assessment of risk of bias in included studies under the domains of sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective outcome reporting and other biases. We judged a study to have a high risk of attrition bias when the reasons for dropouts were not balanced across intervention groups. We judged a study at high risk of bias when at least one high risk of bias among the seven domains existed. Conversly, when a study had low risk of bias for all key domains we judged the study to be at low risk of bias, unless one of the domain was reported as unclear, in this case we judged the study at unclear risk of bias.
Measures of treatment effect
We used rate ratio as the measure of treatment effect for count data, such as the numbers of relapses, new gadolinium‐enhancing T1‐weighted lesions and new or enlarging T2‐weighted hyperintense lesions. The percentage brain volume change was a continuous outcome and we used the mean difference (MD) as the measure of treatment effect. For dichotomous outcomes, such as the proportion of patients with at least one relapse, disability worsening and at least one AE, we used the risk ratio (RR) as the measure of treatment effect. We also calculated pooled statistics as risk difference (RD) (also called the absolute risk reduction) and the number needed to treat to benefit (NNTB) (NNTB=1/RD). We treated the data on HRQoF scales as continuous because they were longer ordinal rating scales and had a reasonably large number of categories. Therefore, we used the MD for trials that used the same rating scale. Where different rating scales were used, we expressed the measure of the treatment difference as the standardised mean difference (SMD).
Unit of analysis issues
Both RCTs on dimethyl fumarate for MS were multi‐arm studies with two dosage of dimethyl fumarate groups (low‐dose: BG‐12 240 mg twice daily; high‐dose: 240 mg three times daily) and a common placebo group, involving repeated observations on participants. All data were presented for each of the groups to which participants were randomised. We created two pair‐wise comparisons of intervention groups to conduct independent meta‐analyses (high‐dose dimethyl fumarate group versus placebo group; low‐dose dimethyl fumarate group versus placebo group). Meanwhile, we performed each separate analysis based on the pre‐set outcomes and different periods of follow‐up.
Dealing with missing data
The data of mean change in HRQoL measured with SF‐36 were incomplete in the published reports, therefore we contacted Biogen Idec Medical Information for unpublished data. When not provided on request, we analysed the available data and performed a sensitivity analysis according to a likely scenario where we assumed that the participants who dropped out both in the experimental group and in the control group had poor outcomes. We addressed the potential impact of missing data on the findings of the review in the Discussion.
Assessment of heterogeneity
We assessed clinical heterogeneity by examining the characteristics of the studies, the similarity between the types of participants, the interventions and the outcomes. We also evaluated the variability in study design and risk of bias (methodological heterogeneity). We evaluated statistical heterogeneity where clinical and methodological heterogeneity were not obvious across the included studies. When pooling trials in meta‐analyses, we calculated the I2 statistic to identify heterogeneity across the two studies. When the I2 value was higher than 30% there was some level of heterogeneity (Higgins 2011). When tests for heterogeneity were statistically significant and inspection of the individual results suggested that it was still logical to combine results, we calculated the overall effects using a random‐effects model.
Assessment of reporting biases
We compared outcomes in the protocol with those in published report where the protocol was available. Otherwise, we compared outcomes listed in the methods section of an article with those whose results were reported. When non‐significant results were mentioned but not reported adequately, there was a possible reporting bias in meta‐analyses. In future updates, and if sufficient RCTs are included in meta‐analysis (≥10 RCTs), we will examine potential publication bias using a funnel plot.
Data synthesis
When clinically and methodologically homogeneous RCTs were identified and heterogeneity tests suggested an I2 value lower than or equal to 30%, or inspection of the individual results suggested that it still seemed logical to combine results even though tests for heterogeneity were statistically significant, we conducted formal meta‐analysis using Review Manager software (Review Manager 2014) (version 5.3.3). We calculated treatment effect estimates for each study and the weighted average of the treatment effects estimated in the individual studies (as a pooled treatment effect estimate), and selected a random‐effects model or fixed‐effect model according to the results of the heterogeneity tests. In the absence of heterogeneity, we used a fixed‐effect model, otherwise we used a random‐effects model. For the outcomes treated as dichotomous data (the proportion of patients with at least one relapse, disability worsening and at least one AE), we used the Mantel‐Haenszel method (Greenland 1985; Mantel 1959). For the outcomes treated as continuous data (the percentage brain volume change and mean change in HRQoF), we used the inverse‐variance method (DerSimonian 1986). For the count data (the ARR, the mean number of gadolinium‐enhancing T1‐weighted lesions and new or enlarging T2‐weighted hyperintense lesions), we used the inverse‐variance method (DerSimonian 1986).
Subgroup analysis and investigation of heterogeneity
We did not conduct subgroup analysis due to a paucity of data. If enough data are available in future updates, we will carry out subgroup analysis according to the following covariates considered as source of heterogeneity.
Different MS patients (e.g. patients with RRMS, patients with progressive MS)
Baseline EDSS scores (e.g. equal to or lower than 3.5, between 3.5 and 6)
Duration of MS (e.g. five years, longer than five years)
Sensitivity analysis
We conducted a sensitivity analysis according to a likely scenario where we assumed that participants with missing outcomes both in the experimental group and in the control group had poor outcomes. Based on the intention‐to‐treat (ITT) principle, we included all randomly assigned patients (including those who did not receive study treatment) into sensitivity analyses.
'Summary of findings' table
We created a 'Summary of findings' table using the following outcomes.
The proportion of patients with at least one relapse at two years of follow‐up
The proportion of patients with disability worsening as assessed by the EDSS at two years of follow‐up
The proportion of patients who discontinued study drug because of AEs at two years of follow‐up
The proportion of patients with lymphopenia at two years of follow‐up
We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of the body of evidence as it related to the studies that contributed data to the meta‐analyses for the prespecified outcomes. We used the methods and recommendations in Section 8.5 and Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) and the GRADEpro software (GRADEpro 2008). We justified all decisions to downgrade or upgrade the quality of studies using footnotes and we made comments to aid the reader's understanding of the review where necessary.
Results
Description of studies
See: "Characteristics of included studies", and "Characteristics of excluded studies"
Results of the search
A total of 137 references were retrieved by the search strategy. After screening of titles and abstracts, 28 references were provisionally selected. The full papers were obtained for further assessment of eligibility. We excluded two studies (reported in four articles): one study was an uncontrolled pilot study (reported in one article) (Schimrigk 2006); the other was a RCT with a length of follow‐up shorter than one year (reported in three articles) (Kappos 2008). Two studies reported in the 24 selected articles were included in the review (Fox 2012; Gold 2012). No ongoing study was eligible according to the inclusion criteria of the review. See Figure 1.
1.
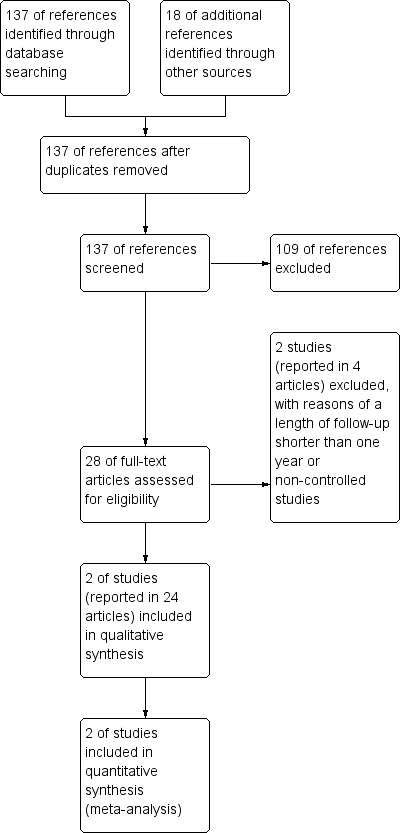
Study flow diagram.
Included studies
Two studies (Fox 2012; Gold 2012) were included, in which 2667 patients were randomly assigned to high‐dose dimethyl fumarate (761), low‐dose dimethyl fumarate (773), placebo (773) and glatiramer acetate (360). Four (0.5%) of 773 patients in the low‐dose dimethyl fumarate group, two (0.3%) of 773 patients in the placebo group and 10 (2.8%) of 360 patients in the glatiramer acetate group did not receive treatment. A subsample of 1221 (45.8%) patients were selected from the randomised population to participate in MRI evaluations by each study site with MRI capabilities itself. Both studies aimed to evaluate the benefit and safety of dimethyl fumarate as monotherapy versus placebo for adults with relapsing‐remitting multiple sclerosis (RRMS). Subcutaneous glatiramer acetate at a dose of 20 mg per day was included as a reference comparator for a relative benefit‐risk assessment of dimethyl fumarate through comparison of the active‐treatment groups with the placebo group in Fox 2012. (Characteristics of included studies).
Characteristics of the study design
Both studies were randomised, double‐blind, placebo‐controlled clinical trial with parallel design for a two‐year follow‐up. Overall, 572 (21.4%) patients withdrew from study: 168 (22.1%) of 761 high‐dose dimethyl fumarate patients, 170 (22.0%) of 773 low‐dose dimethyl fumarate patients, 176 (22.8%) of 773 placebo controls and 58 (16.1%) of 360 glatiramer acetate patients. Not all randomly assigned patients were included in data analyses. Sixteen patients randomised but not receiving study treatment were excluded from the data analyses. For the MRI outcomes, only those with available post baseline MRI data were included in the analyses. Missing MRI data after patients switched to alternative MS medications were imputed with the use of a constant‐rate assumption. The ITT principle was not used.
Characteristics of the participants
Both studies included participants with (1) a diagnosis of RRMS according to the 2005 revision of the "McDonald Criteria" (Polman 2005); (2) an age of 18 to 55 years; (3) a score of 0 to 5 on the EDSS; (4) disease activity as evidenced by at least one clinically documented relapse in the previous 12 months or at least one gadolinium‐enhancing lesion 0 to 6 weeks before randomisation. Patients with progressive forms of MS were excluded. Baseline demographic and disease characteristics were similar among the study groups (including the MRI cohort and the HRQoL cohort).
Characteristics of the interventions
Both studies had multiple arms with two dosages of dimethyl fumarate groups (low‐dose: 240 mg orally twice daily for 96 weeks, high‐dose: 240 mg orally three times daily for 96 weeks) and a common placebo control group. Additionally, a rater‐blinded, active agent approved for RRMS (subcutaneous glatiramer acetate at a dose of 20 mg per day) was included as a reference comparator in Fox 2012.
Characteristics of the outcome measures
Both studies reported the data at two years of follow‐up only, data at one year were not available. The proportion of patients with at least one relapse was reported as the primary outcome in Gold 2012 but as one of the secondary outcomes in Fox 2012. The proportion of patients with disability worsening was reported as one of secondary outcomes in both studies. The annualised relapse rate (ARR) was reported as the primary outcome in Fox 2012. The following outcomes were reported as secondary outcomes in both studies: the proportion of patients with at least one AE, the proportion of patients with at least one SAE, the proportion of patients who discontinued study drug because of AEs, the number (rate) of gadolinium‐enhancing T1‐weighted lesions, the number (rate) of new or enlarging T2‐weighted hyperintense lesions and mean change in HRQoL. The percentage brain volume change was not an outcome measure in either study.
Excluded studies
Two studies were excluded from this review. Reasons for their exclusion are available in the Characteristics of excluded studies table.
Risk of bias in included studies
Further details of this assessment are available in the relevant section of the Characteristics of included studies table and are also presented in the 'Risk of bias' graph (Figure 2) and 'Risk of bias' summary (Figure 3).
2.
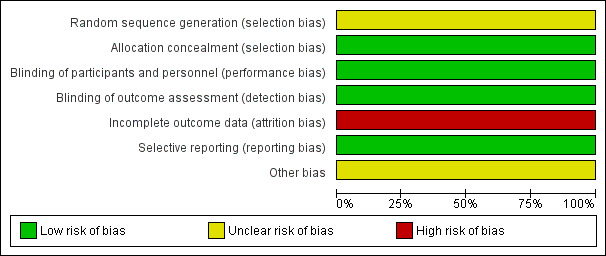
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Allocation
As described in the study protocols, randomisation took place across all study sites using a centralised Interactive Voice Response System (IVRS). The randomisation was stratified by site. At randomisation, the centralised IVRS assigned a unique six‐digit subject identification number to each participant (the first segment of the number represents the study site and the second segment of the number represents the participant at that study site). However, the specific method of random sequence generation was not described. The risk of selection bias was unclear. For the MRI outcomes, there was a high risk of selection bias in both studies because the participants in MRI cohort were selected by each study site with MRI capabilities itself, which led to an obvious heterogeneity between the studies.
Blinding
A double‐blinded design was used in both studies. The participants, personnel and outcome assessors were blinded to the assignment to dimethyl fumarate and placebo. Only the outcome assessors were blinded to the assignment to glatiramer acetate. The risks of performance bias and detection bias were low. However, despite that patients were instructed to take the assigned study drug at least four hours before study visits in order to keep the personnel and investigators unaware of the assignment to dimethyl fumarate, the most frequent side effect of flushing made it likely that patients could become unblinded during the course of the trial.
Incomplete outcome data
A total of 282 (22.8%) patients in Gold 2012 and 290 (20.3%) patients in Fox 2012 withdrew from study over a period of two years. The unbalanced reasons for withdrawal among groups, along with an high overall dropout rate of 21.4%, contributed to a high risk of attrition bias.
Selective reporting
All listed outcomes were reported adequately in the study. The risk of reporting bias was low.
Other potential sources of bias
The study was funded by Biogen Idec. Data were gathered by the investigators and were analysed by the sponsor (Biogen Idec). Conflict of interests may exist.
Effects of interventions
See: Table 1
See: Table 1. There were no available data at the first year of follow‐up.
Primary outcomes
Benefit
(1) The proportion of patients with at least one relapse at two years of follow‐up
Overall, the risk of new relapse in participants receiving high‐dose and low‐dose dimethyl fumarate was 25.10% and 27.96% respectively, significantly lower than that in participants receiving placebo (43.71%), the risk difference (RD) was 18.61% and 15.75% respectively. Compared to placebo, the pooled risk ratio (RR) with high‐dose dimethyl fumarate administration was 0.57 (95% confidence interval (CI) 0.50 to 0.66, P < 0.00001; two studies 1532 participants (Analysis 1.1)), and the NNTB was 5, which means that there need to treat five patients with high‐dose dimethyl fumarate to prevent one patient against relapse during the two years of follow‐up. By contrast, the pooled RR with low‐dose dimethyl fumarate administration was 0.64 (95% CI 0.54 to 0.77, P < 0.00001; two studies 1540 participants (Analysis 2.1)), and the NNTB was 6, which means that there need to treat six patients with low‐dose dimethyl fumarate to prevent one patient against relapse during the two years of follow‐up. Assuming participants who withdrew from study both in experimental groups and control group had a relapse, the likely‐case scenario analysis showed both dosages of dimethyl fumarate reduced the number of patients with relapse at two years of follow‐up (RR = 0.75, 95% CI 0.68 to 0.83, P < 0.00001; two studies 1534 participants (Analysis 1.2) and RR = 0.81, 95% CI 0.74 to 0.89, P < 0.0001; two studies 1546 participants (Analysis 2.2), respectively).
1.1. Analysis.
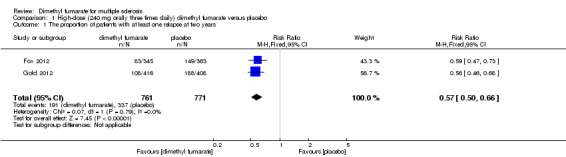
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 1 The proportion of patients with at least one relapse at two years.
2.1. Analysis.
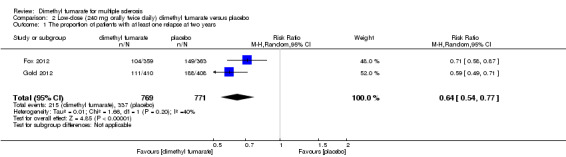
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 1 The proportion of patients with at least one relapse at two years.
1.2. Analysis.
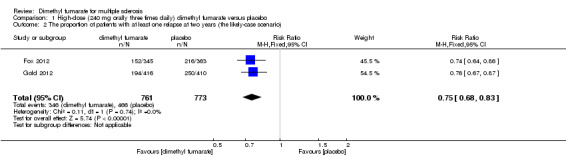
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 2 The proportion of patients with at least one relapse at two years (the likely‐case scenario).
2.2. Analysis.
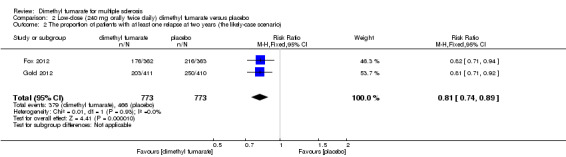
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 2 The proportion of patients with at least one relapse at two years (the likely‐case scenario).
Compared to glatiramer acetate, there was a significant difference in reducing the number of patients with relapse for high dosage of dimethyl fumarate (RR = 0.75, 95% CI 0.59 to 0.96, P = 0.02); but no difference for low dosage (RR = 0.91, 95% CI 0.72 to 1.13, P = 0.38). Taking the effect of dropouts into consideration, there was no difference in the likely‐case scenario analysis (RR = 0.91, 95% CI 0.78 to 1.07, P = 0.26 and RR = 1.01, 95% CI 0.87 to 1.17, P = 0.94, respectively).
(2) The proportion of patients with disability worsening as assessed by the EDSS at two years of follow‐up
Disability worsening was confirmed at least 12 weeks (less than six months) in both studies. Based on such data, the risk of disability worsening in participants receiving high‐dose and low‐dose dimethyl fumarate was 15.77% and 14.58% respectively, which were lower than that in participants receiving placebo (22.31%), the RD was 6.54% and 7.73% respectively. Compared to placebo, the pooled RR with high‐dose dimethyl fumarate administration was 0.70 (95% CI 0.57 to 0.87, P = 0.0009; two studies 1532 participants (Analysis 1.4)), and the NNTB was 15, which means that there need to treat 15 patients with high‐dose dimethyl fumarate to prevent one patient against disability worsening during the two years of follow‐up. By contrast, the pooled RR with low‐dose dimethyl fumarate administration was 0.65 (95% CI 0.53 to 0.81, P = 0.0001; two studies 1539 participants (Analysis 2.4)), and the NNTB was 13, which means that there need to treat 13 patients with low‐dose dimethyl fumarate to prevent one patient against disability worsening during the two years of follow‐up. Assuming all dropouts from experimental groups and control group had disability worsening, the likely‐case scenario analysis showed both dosages of dimethyl fumarate reduced the number of patients with disability worsening at two years of follow‐up (RR = 0.85, 95% CI 0.75 to 0.97, P = 0.01; two studies 1534 participants (Analysis 1.5) and RR = 0.83, 95% CI 0.73 to 0.94, P = 0.004; two studies 1546 participants (Analysis 2.5), respectively).
1.4. Analysis.
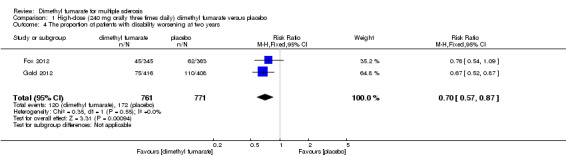
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 4 The proportion of patients with disability worsening at two years.
2.4. Analysis.
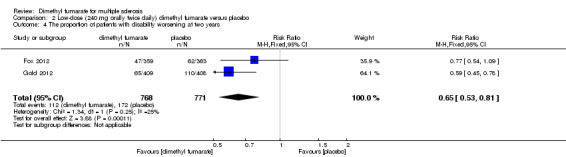
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 4 The proportion of patients with disability worsening at two years.
1.5. Analysis.
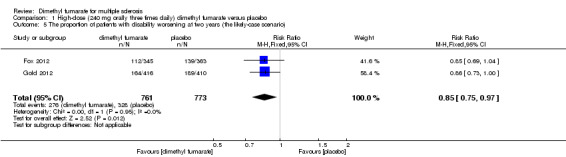
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 5 The proportion of patients with disability worsening at two years (the likely‐case scenario).
2.5. Analysis.
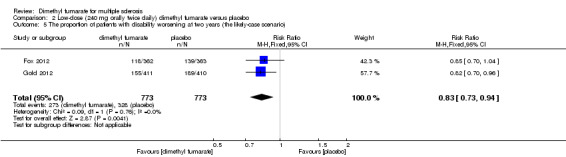
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 5 The proportion of patients with disability worsening at two years (the likely‐case scenario).
Compared to glatiramer acetate, there was no significant difference in reducing the number of patients with disability worsening for both dosages of dimethyl fumarate (high dosage: RR = 0.82, 95% CI 0.57 to 1.17, P = 0.27; low dosage: RR = 0.82, 95% CI 0.57 to 1.17, P = 0.27). Taking the effect of dropouts into consideration, there was no difference in the likely‐case scenario in sensitivity analysis (RR = 1.01, 95% CI 0.81 to 1.25, P = 0.95 and RR = 1.01, 95% CI 0.82 to 1.25, P = 0.91, respectively).
Safety
(1) The number of patients with at least one adverse event (AE) at two years of follow‐up
Overall, compared with placebo group, the pooled results showed that the incidence of AEs excluding relapses was significantly increased by both dosages of dimethyl fumarate administration (high dosage: RR = 1.38, 95% CI 1.27 to 1.51, P < 0.00001; two studies 1531 participants (Analysis 1.8); low dosage: RR = 1.37, 95% CI 1.25 to 1.49, P < 0.00001; two studies 1540 participants (Analysis 2.8), respectively). The most common AEs included flushing (high dosage: RR = 6.57, 95% CI 4.62 to 9.35, P < 0.00001; two studies 1531 participants (Analysis 1.11); low dosage: RR = 8.01, 95% CI 5.66 to 11.34, P < 0.00001; two studies 1540 participants (Analysis 2.11)); upper abdominal pain (RR = 1.91, 95% CI 1.35 to 2.69, P = 0.0003; two studies 1531 participants (Analysis 1.12) and RR = 1.69, 95% CI 1.19 to 2.41, P = 0.004; two studies 1540 participants (Analysis 2.12), respectively); nausea (RR= 1.59, 95% CI 1.19 to 2.12, P = 0.002); two studies 1531 participants (Analysis 1.13) and RR= 1.39, 95% CI 1.03 to 1.87, P = 0.03; two studies 1540 participants (Analysis 2.13), respectively); diarrhoea (RR = 1.55, 95% CI 1.20 to 2.01, P = 0.0008); two studies 1531 participants (Analysis 1.14) and RR = 1.31, 95% CI 0.91 to 1.87, P = 0.14; two studies 1540 participants (Analysis 2.14), respectively); and proteinuria (RR = 1.46, 95% CI 1.06 to 2.00, P = 0.02; two studies 1531 participants (Analysis 1.15) and RR = 1.14, 95% CI 0.81 to 1.59, P = 0.45; two studies 1540 participants (Analysis 2.15), respectively). Other uncommon AEs associated with dimethyl fumarate administration included lymphopenia (RR = 5.25, 95% CI 2.20 to 12.51, P = 0.0002; two studies 1531 participants (Analysis 1.16) and RR = 5.69, 95% CI 2.40 to 13.46, P < 0.0001; two studies 1540 participants (Analysis 2.16), respectively) and leukopenia (RR = 5.23, 95% CI 2.47 to 11.07, P < 0.0001; two studies 1531 participants (Analysis 1.17) and RR = 6.53, 95% CI 3.13 to 13.64, P < 0.00001; two studies 1540 participants (Analysis 2.17), respectively). There was no difference in the number of patients with an increased alanine aminotransferase level at least three times the upper limit of the normal range at two years (RR = 1.34, 95% CI 0.61 to 2.94, P = 0.46; two studies 1531 participants (Analysis 1.18) and RR = 1.33, 95% CI 0.57 to 3.07, P = 0.51; two studies 1540 participants (Analysis 2.18), respectively). Overall, the incidence of gastrointestinal events (including upper abdominal pain, nausea and diarrhoea) was increased by both dosages (RR = 1.67, 95% CI 1.31 to 2.12, P < 0.0001; two studies 1531 participants (Analysis 1.19) and RR = 1.43, 95% CI 1.11 to 1.85, P = 0.005; two studies 1540 participants (Analysis 2.19), respectively). In terms of infections, the was no difference either in the number of patients with infections (RR = 1.08, 95% CI 0.99 to 1.17, P = 0.08; two studies 1531 participants (Analysis 1.20) and RR = 1.04, 95% CI 0.92 to 1.18, P = 0.54; two studies 1540 participants (Analysis 2.20), respectively) and in the number of patients with serious infections (RR = 1.38, 95% CI 0.64 to 2.98, P = 0.41; two studies 1531 participants (Analysis 1.21) and RR = 1.55, 95% CI 0.73 to 3.28, P = 0.25; two studies 1540 participants (Analysis 2.21), respectively). No opportunistic infections were reported in any experimental group.
1.8. Analysis.
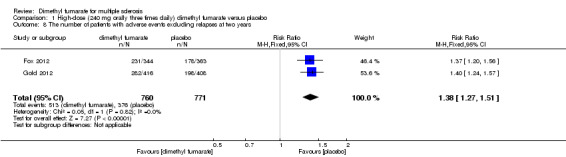
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 8 The number of patients with adverse events excluding relapses at two years.
2.8. Analysis.
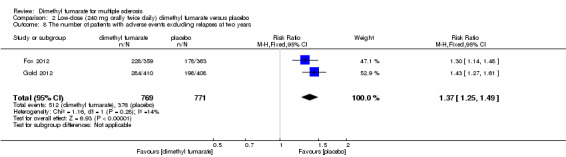
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 8 The number of patients with adverse events excluding relapses at two years.
1.11. Analysis.
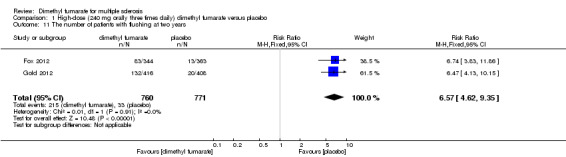
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 11 The number of patients with flushing at two years.
2.11. Analysis.
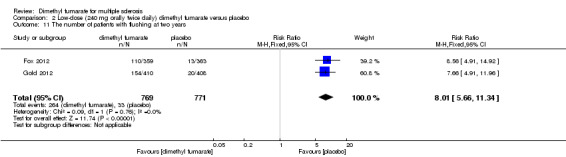
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 11 The number of patients with flushing at two years.
1.12. Analysis.
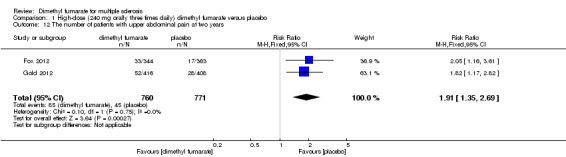
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 12 The number of patients with upper abdominal pain at two years.
2.12. Analysis.
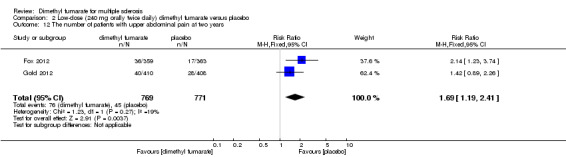
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 12 The number of patients with upper abdominal pain at two years.
1.13. Analysis.
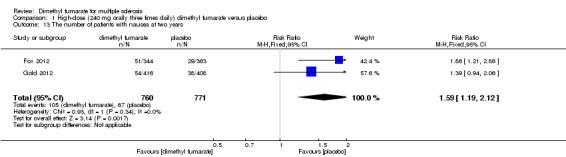
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 13 The number of patients with nausea at two years.
2.13. Analysis.
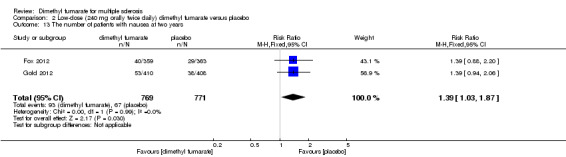
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 13 The number of patients with nausea at two years.
1.14. Analysis.
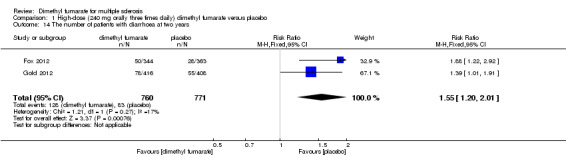
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 14 The number of patients with diarrhoea at two years.
2.14. Analysis.
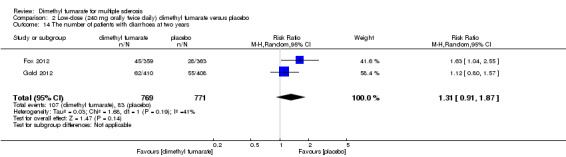
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 14 The number of patients with diarrhoea at two years.
1.15. Analysis.
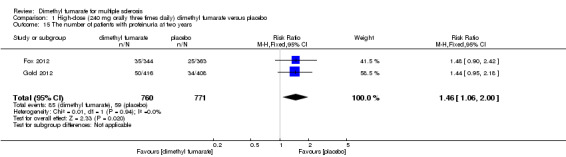
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 15 The number of patients with proteinuria at two years.
2.15. Analysis.
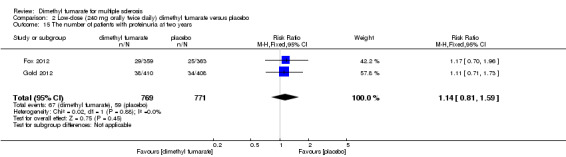
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 15 The number of patients with proteinuria at two years.
1.16. Analysis.
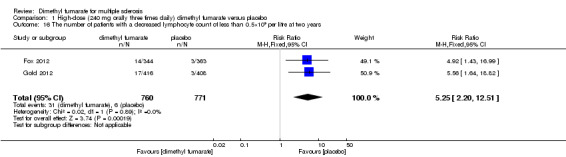
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 16 The number of patients with a decreased lymphocyte count of less than 0.5×109 per litre at two years.
2.16. Analysis.
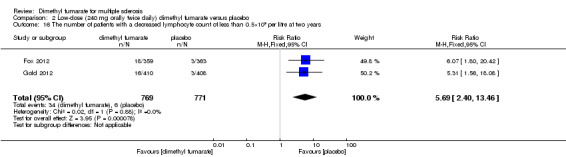
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 16 The number of patients with a decreased lymphocyte count of less than 0.5×109 per litre at two years.
1.17. Analysis.
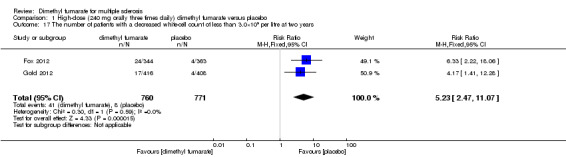
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 17 The number of patients with a decreased white‐cell count of less than 3.0×109 per litre at two years.
2.17. Analysis.
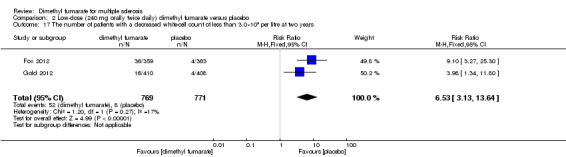
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 17 The number of patients with a decreased white‐cell count of less than 3.0×109 per litre at two years.
1.18. Analysis.
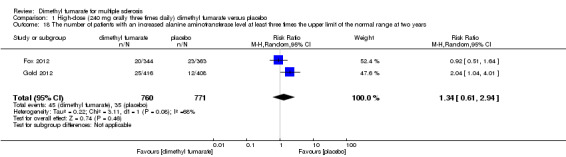
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 18 The number of patients with an increased alanine aminotransferase level at least three times the upper limit of the normal range at two years.
2.18. Analysis.
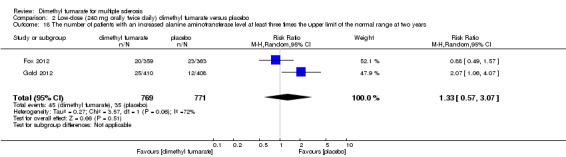
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 18 The number of patients with an increased alanine aminotransferase level at least three times the upper limit of the normal range at two years.
1.19. Analysis.
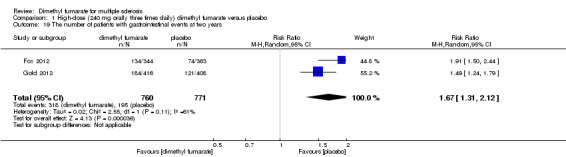
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 19 The number of patients with gastrointestinal events at two years.
2.19. Analysis.
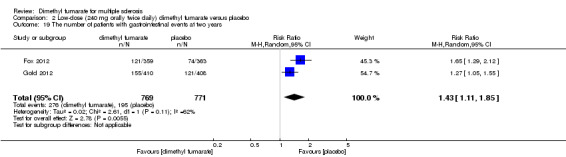
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 19 The number of patients with gastrointestinal events at two years.
1.20. Analysis.
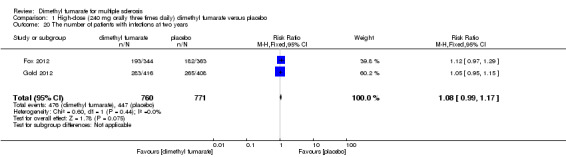
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 20 The number of patients with infections at two years.
2.20. Analysis.
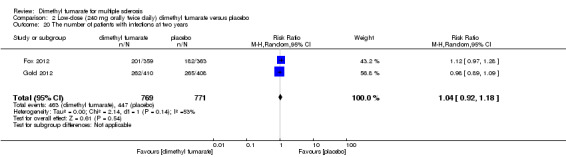
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 20 The number of patients with infections at two years.
1.21. Analysis.
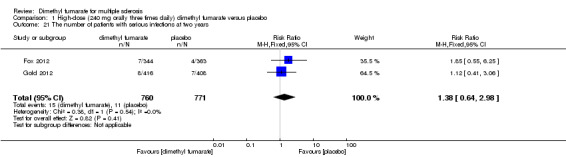
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 21 The number of patients with serious infections at two years.
2.21. Analysis.
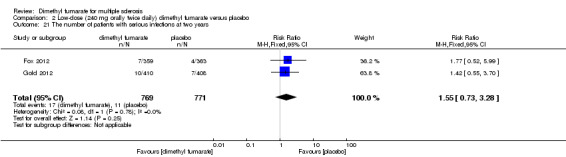
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 21 The number of patients with serious infections at two years.
(2) The number of patients with at least one SAE at two years of follow‐up
The pooled risk of SAEs excluding relapses both in participants receiving high‐dose and low‐dose dimethyl fumarate was not higher than that in participants receiving placebo (RR = 1.07, 95% CI 0.75 to 1.53, P = 0.71; two studies 1531 participants (Analysis 1.9) and RR = 1.05, 95% CI 0.63 to 1.74, P = 0.87; two studies 1540 participants (Analysis 2.9), respectively).
1.9. Analysis.
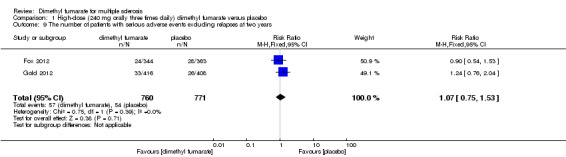
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 9 The number of patients with serious adverse events excluding relapses at two years.
2.9. Analysis.
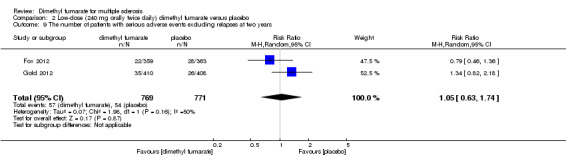
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 9 The number of patients with serious adverse events excluding relapses at two years.
(3) The number of patients who discontinued study drug because of AEs at two years of follow‐up
There was a significant difference in the number of patients who discontinued study drug because of AEs excluding relapses between participants receiving dimethyl fumarate and participants receiving placebo (high dosage: RR = 2.16 (95% CI 1.54 to 3.03, P < 0.00001); two studies 1531 participants (Analysis 1.10); low dosage: RR = 2.18 (95% CI 1.56 to 3.06, P < 0.00001; two studies 1540 participants (Analysis 2.10)). Overall, the incidences of study drug discontinuation due to adverse effects both in high‐dose group and low‐dose group, such as diarrhoea (1.97% and 0.91%, respectively), flushing (1.58% and 3.12%, respectively), nausea (1.58% and 0.78%, respectively) and upper abdominal pain (1.32% and 0.78%, respectively) were low.
1.10. Analysis.

Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 10 The number of patients who discontinued study drug because of adverse events excluding relapses at two years.
2.10. Analysis.
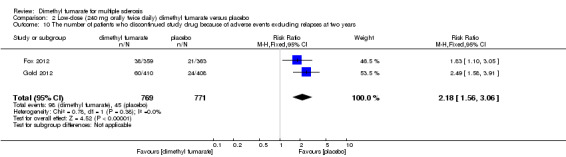
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 10 The number of patients who discontinued study drug because of adverse events excluding relapses at two years.
Secondary outcomes
(1) The ARR at two years of follow‐up
Compared to placebo, the pooled results showed both dosages of dimethyl fumarate significantly reduced the ARR at two years of follow‐up (high dosage: rate ratio = 0.51, 95% CI 0.45 to 0.59, P < 0.00001; two studies 1532 participants (Analysis 1.3); low dosage: rate ratio = 0.51, 95% CI 0.44 to 0.59, P < 0.00001; two studies 1540 participants (Analysis 2.3)) .
1.3. Analysis.
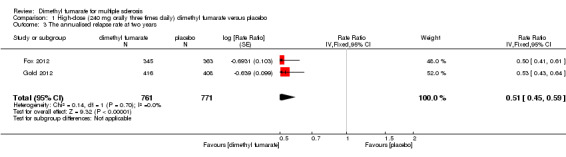
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 3 The annualised relapse rate at two years.
2.3. Analysis.
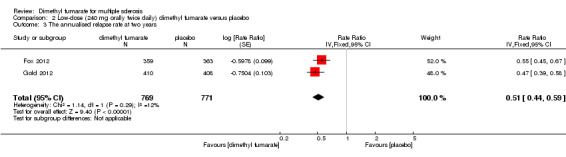
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 3 The annualised relapse rate at two years.
Compared to glatiramer acetate, there was a significant difference in reducing the ARR for both dosages (high dosage: rate ratio = 0.69, 95% CI 0.56 to 0.86, P = 0.0007; low dosage: rate ratio = 0.76, 95% CI 0.62 to 0.94, P = 0.01).
(2) The number of gadolinium‐enhancing T1‐weighted lesions at two years of follow‐up
We did not conduct meta‐analyses for the data of active lesions on MRI scans as there was a high risk of selection bias for MRI outcomes in both studies, which led to an obvious heterogeneity between the studies. Moreover, MRI data reported in both studies showed imprecision because an improper method was used to handle the missing data. We calculated the rate ratio using the available MRI data in each study.
Compared to placebo, there was a significant difference in reducing the number of gadolinium‐enhancing T1‐weighted lesions for both dosages of dimethyl fumarate in Gold 2012 (high dosage: rate ratio = 0.28, 95% CI 0.22 to 0.36, P < 0.00001; low dosage: rate ratio = 0.06, 95% CI 0.03 to 0.09, P < 0.00001), as well as in Fox 2012 (high dosage: rate ratio = 0.25, 95% CI 0.19 to 0.32, P < 0.00001; low dosage: rate ratio = 0.25, 95% CI 0.19 to 0.32, P < 0.00001). Compared to glatiramer acetate, there was a significant difference in reducing the number of gadolinium‐enhancing T1‐weighted lesions for both dosages of dimethyl fumarate (high dosage: rate ratio = 0.57, 95% CI 0.41 to 0.78, P = 0.0005; low dosage: rate ratio = 0.71, 95% CI 0.53 to 0.95, P = 0.02).
(3) The number of new or enlarging T2‐weighted hyperintense lesions at two years of follow‐up
Compared to placebo, there was a significant difference in reducing the number of new or enlarging T2‐weighted hyperintense lesions for both dosages of dimethyl fumarate in Gold 2012 (high dosage: rate ratio = 0.26, 95% CI 0.24 to 0.28, P < 0.00001; low dosage: rate ratio = 0.15, 95% CI 0.14 to 0.17, P < 0.00001), as well as in Fox 2012 (high dosage: rate ratio = 0.27, 95% CI 0.25 to 0.29, P < 0.00001; low dosage: rate ratio = 0.29, 95% CI 0.27 to 0.32, P < 0.00001). Compared to glatiramer acetate, there was a significant difference in reducing the number of new or enlarging T2‐weighted hyperintense lesions for both dosages of dimethyl fumarate (high dosage: rate ratio = 0.59, 95% CI 0.54 to 0.65, P < 0.00001; low dosage: rate ratio = 0.64, 95% CI 0.58 to 0.70, P < 0.00001).
(4) Mean change in HRQoL measured with SF‐36 from baseline to two years
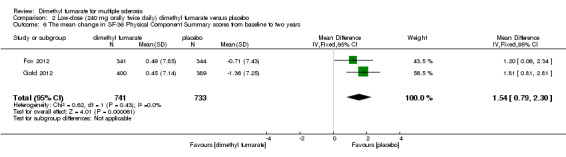
Data from two studies with a total of 1461 patients were available to analyse the mean change in HRQoL measured with SF‐36 from baseline to two years. Compared to placebo, the pooled results showed treatment with dimethyl fumarate slightly improved the physical HRQoL measured by mean change in SF‐36 Physical Component Summary (PCS) scores from baseline to two years (high dosage: mean difference (MD) = 1.51, 95% CI 0.76 to 2.26, P < 0.0001; two studies 1461 participants (Analysis 1.6); low dosage: MD = 1.54, 95% CI 0.79 to 2.30, P < 0.0001; two studies 1474 participants (Analysis 2.6)), but did not improve the mental HRQoL measured by mean change in SF‐36 Mental Component Summary (MCS) scores from baseline to two years (high dosage: MD = 1.19, 95% CI ‐ 0.70 to 3.08, P = 0.22; two studies 1461 participants (Analysis 1.7); low dosage: MD = 0.93, 95% CI ‐ 0.06 to 1.93, P = 0.07; two studies 1474 participants (Analysis 2.7)).
1.6. Analysis.
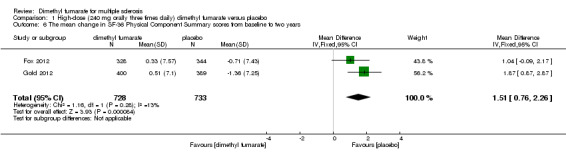
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 6 The mean change in SF‐36 Physical Component Summary scores from baseline to two years.
2.6. Analysis.
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 6 The mean change in SF‐36 Physical Component Summary scores from baseline to two years.
1.7. Analysis.
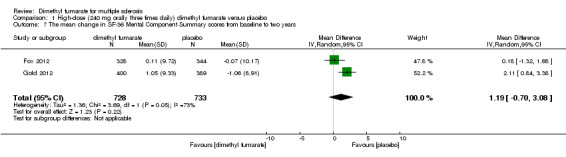
Comparison 1 High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo, Outcome 7 The mean change in SF‐36 Mental Component Summary scores from baseline to two years.
2.7. Analysis.
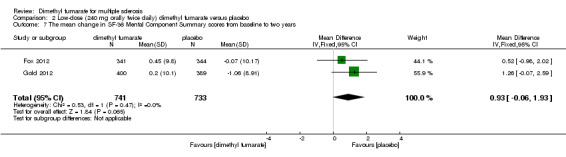
Comparison 2 Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo, Outcome 7 The mean change in SF‐36 Mental Component Summary scores from baseline to two years.
Discussion
Summary of main results
This systematic review included only two randomised controlled trials (RCTs) involving 2667 adult patients with relapsing‐remitting multiple sclerosis (RRMS). All participants had a score of zero to five on the Expanded Disability Status Scale (EDSS) and disease activity with at least one relapse in the previous 12 months or at least one gadolinium‐enhancing lesion during the previous six weeks. Both studies aimed to evaluate the benefit of dimethyl fumarate as monotherapy versus placebo in respect of relapse, disability worsening, brain lesions, HRQoF and the safety over a period of two years. One RCT was excluded because of the length of follow‐up shorter than one year. Both studies included in this review were reported in multiple publications. The characteristics including study design, types of participants, interventions and outcome measures were similar. There were no obvious clinical and methodological heterogeneities between studies. However, both studies had a high risk of attrition bias resulting from a high overall dropout rate and the unbalanced reasons for withdrawal among the groups over the two years of follow‐up, combined with failure to include all randomised patients into intention‐to‐treat analysis.
Compared to placebo, administration of dimethyl fumarate at a dose of 240 mg orally three times daily or twice daily significantly reduced the number of patients with relapse or disability worsening by two years. The likely‐case scenario in sensitivity analysis showed decreased, but still significant, treatment effects on the two outcomes for both dosages. The benefit of both dosages were similar for both outcomes. Compared to placebo, there was also a similar treatment effect on AAR for both dosages. Treatment with both dimethyl fumarate slightly improved the physical HRQoF compared to placebo, but did not improve the mental aspects of HRQoF measured by 36‐SF. Compared to glatiramer acetate, when taking the effect of dropouts into consideration, there was no difference in reducing the number of patients with relapse or disability worsening for both dosages of dimethyl fumarate, however, these results, along with the statistical effect of the ARR, were unconvincing due to a lack of power. We did not conduct meta‐analyses for the MRI outcomes because there was a high risk of selection bias for MRI outcomes and imprecision of MRI data in both studies, as well as an obvious heterogeneity between the studies. The statistical results on MRI outcomes in both studies were unconvincing.
Overall, patients in the placebo group tended to relapse more frequently, which accounted for a large proportion of AEs in the placebo group. After excluding relapses, the risks for other AEs in participants receiving dimethyl fumarate were slightly higher than that in participants receiving placebo, but the risks for study drug discontinuation due to other AEs were increased by both dosages of dimethyl fumarate administration. The most common AEs included flushing, upper abdominal pain, nausea and diarrhoea. The incidence of these events was highest in the first month of treatment, decreasing thereafter. Uncommon AEs, but associated with dimethyl fumarate administration included lymphopenia and leukopenia. Despite this, dimethyl fumarate administration did not increase the incidence of infections and serious infections, it should be noted that patients taking dimethyl fumarate may risk developing a rare and serious brain infection called progressive multifocal leukoencephalopathy (PML) (Ermis 2013, Sweetser 2013, van Oosten 2013). Most recently, the U.S. Food and Drug Administration (FDA) is warning that a patient with MS who was being treated with Tecfidera™ (dimethyl fumarate), developed PML, and later died. As a result, information describing this case of PML, or PML, is being added to the Tecfidera™ drug label (U.S. Food and Drug Administration 2014).
Overall completeness and applicability of evidence
In this review, we excluded one RCT due to length of follow‐up shorter than one year. Generally, disease‐modifying therapy for MS needs an adequate administration duration and follow‐up to determinate the benefit and safety outcomes accurately. A minimum duration of administration of one year, pre‐defined in the criteria of types of interventions, was a reasonable treatment length that partly avoided the inclusion of misleading evidence. There was no study targeting other phenotypes of MS. Limited data were derived from two RCTs specific to patients with RRMS. We performed meta‐analyses according to two different administration frequencies using the available data from such patients. The evidence in this review is only applicable to adult individuals with RRMS, who has a score of zero to five on the EDSS and disease activity with at least one relapse in the previous 12 months or at least one gadolinium‐enhancing lesion during the previous six weeks .
Quality of the evidence
We included two RCTs in this review, involving 2667 adult patients with RRMS to mainly evaluate the benefit and safety of two dosages of dimethyl fumarate (240 mg orally three times daily or twice daily) by direct comparison with placebo. Overall, there were no obvious clinical and methodological heterogeneities between the studies. Both studies had a high attrition bias, resulting in moderate‐quality evidence for most primary outcomes. The results of disability worsening were additionally subjected to a serious indirectness of evidence because disability worsening was confirmed in less than six months in both studies. All these factors contributed to a low quality of the evidence for disability worsening. The quality of MRI data reported in the primary studies was poor.
Potential biases in the review process
An extensive, comprehensive search was undertaken to limit bias in the review process. The two authors' independent assessments of the eligibility of studies for inclusion in this review and the extraction of data minimised the potential for additional bias beyond that detailed in the 'Risk of bias' tables. The authors of this review had no conflicts of interest.
Agreements and disagreements with other studies or reviews
Recently a systematic review (Kawalec 2014) included all available RCTs to evaluate the effectiveness of dimethyl fumarate monotherapy in the treatment of RRMS. The authors included three studies in qualitative synthesis. Among them, two studies, included in the current systematic review as well, were eligible for meta‐analyses. The authors did not fully consider the potential influences of bias on the internal validity of the results and did not conduct a comprehensive analysis for the quality of evidence. In the current review, we evaluated the impact of the five GRADE factors (limitations in study design or execution (risk of bias), inconsistency in results, imprecision of results, indirectness of evidence, publication bias) on the quality of evidence for the outcomes.
Authors' conclusions
Implications for practice.
There is moderate‐quality evidence to support that dimethyl fumarate at a dose of 240 mg orally three times daily or twice daily reduces both on the number of patients with a relapse and the annualised relapse rate over two years of treatment. However, the quality of the evidence to support the benefit in reducing the number of patients with disability worsening is low. There is no high‐quality data available to evaluate the benefit on MRI outcomes. The common adverse effects (AEs) such as flushing and gastrointestinal events (e.g. diarrhoea, nausea, and upper abdominal pain) are mild‐to‐moderate for most of patients. Lymphopenia and leukopenia are uncommon AEs but significantly associated with dimethyl fumarate. Both dosages of dimethyl fumarate have similar benefit and safety profile, which supports the option of low‐dose administration.
Implications for research.
The ideal target of disease‐modifying therapy for MS is to prevent disability worsening and improve quality of life, which are two key aspects generally needed to be considered when evaluating and deciding whether a disease‐modifying drug has superior benefit. Therefore, new studies of high quality and longer follow‐up are needed to evaluate the benefit of dimethyl fumarate on these outcomes and to observe the long‐term adverse effects including progressive multifocal leukoencephalopathy (PML).
Acknowledgements
We thank Andrea Fittipaldo, Trials Search Co‐ordinator, and Liliana Coco, Managing Editor of the Cochrane Multiple Sclerosis and Rare Diseases of the Central Nervous System Group, for their help and support in developing this review. We thank all peer reviewers, and Graziella Filippini, the Co‐ordinating editor of the Cochrane Multiple Sclerosis and Rare Diseases of the Central Nervous System Group, for their constructive comments and suggestions for this review.
Appendices
Appendix 1. Keywords
{dimethyl fumarate} OR {Fumaderm} OR {FAG 201} OR {FAG201} OR {FAG‐201} OR {BG 00012} OR {BG00012} OR {BG‐00012} OR {BG 12 compound} OR {BG12 compound} OR {BG‐12 compound} OR {BG‐12} OR {tecfidera} OR {Nrf2 activator} OR {oral fumarate} OR {fumaric acid eaters}
Data and analyses
Comparison 1. High‐dose (240 mg orally three times daily) dimethyl fumarate versus placebo.
Comparison 2. Low‐dose (240 mg orally twice daily) dimethyl fumarate versus placebo.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Fox 2012.
| Methods | The trial was a randomised, double‐blind, placebo‐controlled phase 3 study. Follow‐up period: 2 years. Population included in data analyses: all randomly assigned patients who received study treatment. For the MRI outcomes, patients with available postbaseline MRI data were included in the analyses. Missing MRI data after patients switched to alternative MS medications were imputed with the use of a constant‐rate assumption. The intention‐to‐treat analysis principle was not used. |
|
| Participants | Inclusion criteria: patients with (1) a diagnosis of RRMS according to the 2005 revisions to the "McDonald Criteria"; (2) aged 18 to 55 years; (3) a score of 0 to 5 on the EDSS; (4) at least one clinically documented relapse in the previous 12 months or at least one gadolinium‐enhancing lesion 0 to 6 weeks before randomisation. Key exclusion criteria: (1) progressive forms of MS; (2) other clinically significant illness, prespecified laboratory abnormalities; (3) prior exposure to glatiramer acetate or contraindicated medications. 1430 patients were randomly assigned to receive four different interventions.Three patients in twice‐daily BG‐12 group and 10 patients in glatiramer acetate did not receive treatment. A subset of 681 (47.6%) patients were selected from the randomised population to participate in MRI evaluations by each study site with MRI capabilities itself. Baseline demographic and disease characteristics were similar among the four study groups. Summary of patient characteristics at baseline (Placebo: G1 (n = 363), Twice‐Daily BG‐12: G2 (n = 359), Thrice‐Daily BG‐12: G3 (n = 345), Glatiramer Acetate: G4 (n = 350)): Age: G1 = 36.9 ± 9.2 years, G2 = 37.8 ± 9.4 years, G3 = 37.8 ± 9.4 years, G3 = 36.7 ± 9.1 years Sex: female G1 = 251 (69%), G2 = 245 (68%), G3 = 250 (72%), G4 = 247 (71%) Weight: G1 = 72.6 ± 16.9 kg, G2 = 71.9 ± 17.9 kg, G3 = 72.5 ± 17.8 kg, G4 = 71.4 ± 19.1 kg Race: White G1 = 305 (84%), G2 = 304 (85%), G3 = 292 (85%), G4 = 290 (83%); Asian G1 = 28 (8%), G2 = 28 (8%), G3 = 26 (8%), G4 = 25 (7%); Black G1 = 9 (2%), G2 = 2 (<1%), G3 = 5 (1%), G4 = 11 (3%); Other or unknown G1 = 21 (6%), G2 = 25 (7%), G3 = 22 (6%), G4 = 24 (7%) Time since diagnosis: G1 = 4.8 ± 5.0 years, G2 = 4.9 ± 5.1 years, G3 = 4.6 ± 5.2 years, G4 = 4.4 ± 4.7 years Any prior approved DMT: G1 = 111 (31%), G2 = 101 (28%), G3 = 100 (29%), G4 = 103 (29%) Relapse in previous 12 months: G1 = 1.4 ± 0.8, G2 = 1.3 ± 0.6, G3 = 1.4 ± 0.7, G4 = 1.4 ± 0.6 EDSS score at baseline: 0 G1 = 13 (4%), G2 = 15 (4%), G3 = 15 (4%), G4 = 18(5%); 1.0 or 1.5 G1 = 78 (21%), G2 = 85 (24%), G3 = 84 (24%), G4 = 77 (22%); 2.0 or 2.5 G1 = 111 (31%), G2 = 94 (26%), G3 = 94 (27%), G4 = 96 (27%); 3.0 or 3.5 G1 = 98 (27%), G2 = 105 (29%), G3 = 99 (29%), G4 = 99 (28%); 4.0 or 4.5 G1 = 50 (14%), G2 = 47 (13%), G3 = 42 (12%), G4 = 46 (13%); 5.0 G1 = 13 (4%), G2 = 12 (3%), G3 = 11 (3%), G4 = 14 (4%) Mean score on EDSS: G1 = 2.6 ± 1.2, G2 = 2.6 ± 1.2, G3 = 2.5 ± 1.2, G4 = 2.6 ± 1.2 |
|
| Interventions | Experimental group1: two 120 mg BG‐12 capsules orally twice daily (BID) and two placebo capsules orally once daily (QD) Experimental group2: two 120 mg BG‐12 capsules orally three times daily (TID) Control group: two placebo capsules orally three times daily (TID) Active Comparator: glatiramer acetate (GA) 20 mg subcutaneous injection once daily (QD) |
|
| Outcomes | The primary outcome measures: the annualised relapse rate at 2 years. The secondary outcome measures: (1) the number of new or enlarging hyperintense lesions on T2‐weighted images at 2 years; (2) the number of new hypointense lesions on T1‐weighted images at 2 years; (3) the proportion of patients with a relapse at 2 years; (4) the time to disability worsening at 2 years. The tertiary outcome measures: (1) a comparison of the relative benefits and risks of BG‐12 or glatiramer acetate versus placebo; (2) the number of gadolinium‐enhancing lesions at 2 years. A relapse was defined as new or recurrent neurologic symptoms not associated with fever or infection, lasting at least 24 hours, accompanied by new objective neurologic findings, and separated from the onset of other confirmed relapses by at least 30 days. Disability worsening was defined as an increase in the EDSS score of at least 1.0 point in patients with a baseline score of 1.0 or more or an increase of at least 1.5 points in patients with a baseline score of 0, confirmed at least 12 weeks later. |
|
| Notes | Patients could switch to an alternative medication for MS if they had two confirmed relapses and had completed 48 weeks of study treatment or if they had confirmed disability worsening. This study was not designed to test the superiority or non‐inferiority of BG‐12 to glatiramer acetate. The study was funded by Biogen Idec. Data were gathered by the investigators and were analysed by the sponsor (Biogen Idec). ClinicalTrials.gov number, NCT00451451. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients at 200 sites in 28 countries were randomly assigned in a 1:1:1:1 ratio to receive oral placebo, BG‐12 at a dose of 240 mg two times daily, BG‐12 at a dose of 240 mg three times daily, or subcutaneous daily injections of 20 mg of glatiramer acetate for 96 week." "At randomisation, the centralized Interactive Voice Response System (IVRS) assigned a unique 6‐digit subject identification number to each subject (the first segment of the number represents the study site and the second segment of the number represents the subject at that study site)." Comment: The authors did not mention the specific method of random sequence generation. We wrote to Biogen Idec Medical Information (medinfo@biogenidec.com) for the methodological information. The staff provided us with the study protocol, but the specific method of random sequence generation was also not reported in the study protocol. The risk of selection bias was unclear. Participants in the MRI cohort were selected from the randomised population by each study site with MRI capabilities itself, the risk of selection bias was high for MRI outcomes. |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomization took place across all study sites using a centralized IVRS. The randomisation was stratified by site." Comment: The method of allocation concealment was not mentioned in the published article. We wrote to Biogen Idec Medical Information (medinfo@biogenidec.com) for the methodological information. The staff provide us with the study protocol. The risk of selection bias was low. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Patients receiving glatiramer acetate were aware of their treatment assignment. All study management and site personnel, investigators, and patients were unaware of assignment to the BG‐12 and placebo groups; examining neurologists, technicians at the magnetic resonance imaging (MRI) reading center, and members of the independent neurologic evaluation committee were unaware of all study‐group assignments. To ensure that the assignments to the BG‐12 and placebo groups would not be revealed, patients in those groups were instructed not to take the study medication within 4 hours before each study visit, since a flushing reaction is known to be more common with BG‐12." Comment: The participants and personnel in this study were blinded to the assignment to the BG‐12 and placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Each site used separate examining and treating neurologists, thereby maintaining rater blinding for all study groups, including the group that received glatiramer acetate." Comment: The outcomes assessors in this study were blinded to all allocated interventions |
| Incomplete outcome data (attrition bias) All outcomes | High risk | According to the Figure S2 (Patient Disposition) shown in Supplementary Appendix, 72 (20.9%) patients in Thrice‐Daily BG‐12 group, 75 (20.7%) in twice‐daily BG‐12 group, 85 (23.4%) in placebo group and 58 (16.1%) in glatiramer acetate group withdrew from study. Comment: To a large degree, missing data and reasons were not balanced among groups. The high overall dropout rate of 20.3% over a period of 2 years had potential impact on the results. Generally, the higher the ratio of participants with missing data to participants with events, the greater potential there is for bias, especially for the high frequency of events. The potential impact of missing continuous outcomes increases with the proportion of participants with missing data. Both MS relapse and the occurrence of adverse events are common events, a high risk of attrition bias existed. |
| Selective reporting (reporting bias) | Low risk | All listed outcomes were reported adequately. The risk of reporting bias was low. |
| Other bias | Unclear risk | Quote: " Data were gathered by the investigators and were analysed by the sponsor (Biogen Idec). The first draft was co‐written by the first and last authors (the latter is a representative of the sponsor), with assistance from a medical‐communications agency paid by the sponsor." Comment: Conflicts of interest may exist. |
Gold 2012.
| Methods | The trial was a randomised, double‐blind, placebo‐controlled phase 3 study. Follow‐up period: 2 years. Population included in data analyses: all efficacy analyses were performed on data from the patients who underwent randomisation and who received at least one dose of the study drug. For the MRI outcomes, patients with available postbaseline MRI data were included in the analyses. Missing MRI data after patients switched to alternative MS medications were imputed with the use of a constant‐rate assumption. The intention‐to‐treat analysis principle was not used. |
|
| Participants | Inclusion criteria: patients with (1) an age of 18 to 55 years; (2) a diagnosis of RRMS as defined according to the 2005 revisions to the "McDonald Criteria" ; (3) a baseline score of 0 to 5.0 on the EDSS;(4) disease activity as evidenced by at least one clinically documented relapse within 12 months before randomisation or a brain magnetic resonance imaging (MRI) scan, obtained within 6 weeks before randomisation,that showed at least one gadolinium‐enhancing lesion. Key exclusion criteria: (1) progressive forms of MS; (2) another major disease that would preclude participation in a clinical trial; (3) abnormal results on prespecified laboratory tests; (4) recent exposure to contraindicated medications. 1237 patients were randomly assigned to receive three different interventions.Two patients in placebo and one in twice‐daily BG‐12 group did not receive treatment. A subset of 540 (43.7%) patients were selected from the randomised population to participate in MRI evaluations by each study site with MRI capabilities itself. All the baseline characteristics were well balanced among the three study groups. Summary of patient characteristics at baseline (Placebo: G1 (n = 408), Twice‐Daily BG‐12: G2 (n = 410), Thrice‐Daily BG‐12: G3 (n = 416)): Age: G1 = 38.5 ± 9.1 years, G2 = 38.1 ± 9.1 years, G3 = 38.8 ± 8.8 years Sex: female G1 = 306 (75%), G2 = 296 (72%), G3 = 306 (74%) Weight: G1 = 71.1 ± 17.0 kg, G2 = 70.7 ± 18.5 kg, G3 = 71.3 ± 16.9 kg Race: White G1 = 318 (78%), G2 = 321 (78%), G3 = 330 (79%); Asian G1 = 42 (10%), G2 = 38 (9%), G3 = 36 (9%); Black G1 = 8 (2%), G2 = 8 (2%), G3 = 10 (2%); Other or unknown G1 = 40 (10%), G2 = 43 (10%), G3 = 40 (10%) Previous use of approved medication for MS: G1 = 172 (42%), G2 = 162 (40%), G3 = 168 (40%) Time since diagnosis: G1 = 5.8 ± 5.8 years, G2 = 5.6 ± 5.4 years, G3 = 5.1 ± 5.3 years Relapse in previous 12 months: G1 = 1.3±0.7, G2 = 1.3±0.7, G3 = 1.3±0.6 EDSS score at baseline: 0 G1 = 21 (5%), G2 = 29 (7%), G3 = 24 (6%); 1.0 or 1.5 G1 = 105 (26%), G2 = 109 (27%), G3 = 104 (25%); 2.0 or 2.5 G1 = 112 (27%), G2 = 116 (28%), G3 = 146 (35%); 3.0 or 3.5 G1 = 97 (24%), G2 = 82 (20%), G3 = 85 (20%); 4.0 or 4.5 G1 = 56 (14%), G2 = 56 (14%), G3 = 42 (10%); 5.0 G1 = 16 (4%), G2 = 16 (4%), G3 = 14 (3%) Mean score on EDSS: G1 = 2.48 ± 1.24, G2 = 2.40 ± 1.29, G3 = 2.36 ± 1.19 Gadolinium‐enhancing T1‐weighted lesions: G1 = 1.6 ± 3.4, G2 = 1.2 ± 3.3, G3 = 1.2 ± 4.1 Hyperintense T2‐weighted lesions: mean G1 = 49.2 ± 38.6, G2 = 47.6 ± 34.7, G3 = 55.8 ± 44.3 The number of patients with < 9 hyperintense T2‐weighted lesions: G1 = 12, G2 = 9, G3 = 14 The number of patients with ≥ 9 hyperintense T2‐weighted lesions: G1 = 168, G2 = 167, G3 = 170 |
|
| Interventions | Experimental group1: two 120 mg BG‐12 capsules orally twice daily (BID) and two 120 mg placebo capsules orally once daily (QD) Experimental group2: two 120 mg BG‐12 capsules orally three times daily (TID) Control group: two 120 mg placebo capsules orally three times daily (TID) |
|
| Outcomes | The primary outcome measures: the proportion of patients who had a relapse by 2 years The secondary outcome measures: (1) the number of gadolinium‐enhancing lesions at 2 years; (2) the number of new or enlarging hyperintense lesions on T2‐weighted images at 2 years; (3) the annualised relapse rate at 2 years (excluding data obtained after patients switched to alternative MS medications); (4) the time to worsening of disability. A relapse was defined as new or recurrent neurologic symptoms, not associated with fever or infection, that lasted for at least 24 hours and that were accompanied by new objective neurologic findings according to the examining neurologist's evaluation. Disability worsening was defined as at least a 1.0‐point increase on the EDSS in patients with a baseline score of 1.0 or higher or at least a 1.5‐point increase in patients with a baseline score of 0, with the increased score sustained for at least 12 weeks. |
|
| Notes | All patients were permitted to switch to an approved alternative MS therapy if they had completed 48 weeks of blinded treatment and experienced at least 1 confirmed relapse after 24 weeks, or at any time if they had experienced disability worsening sustained for 12 weeks. The study was funded by Biogen Idec. Data were collected by the investigators and were analysed by the sponsor (Biogen Idec). ClinicalTrials.gov number, NCT00420212. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients at 198 sites in 28 countries were randomly assigned, in a 1:1:1 ratio, to receive, in a double‐blind fashion, BG‐12 at a dose of 240 mg twice daily, BG‐12 at a dose of 240 mg three times daily, or placebo." "At randomisation, the centralized Interactive Voice Response System (IVRS) assigned a unique 6‐digit subject identification number to each subject (the first segment of the number represents the study site and the second segment of the number represents the subject at that study site)." Comment: The authors did not mention the specific method of random sequence generation. We wrote to Biogen Idec Medical Information (medinfo@biogenidec.com) for the methodological information. The staff provided us with the study protocol, but the specific method of random sequence generation was also not reported in the study protocol. The risk of selection bias was unclear. Participants in the MRI cohort were selected from the randomised population by each study site with MRI capabilities itself, the risk of selection bias was high for MRI outcomes. |
| Allocation concealment (selection bias) | Low risk | Quote: "Randomisation took place across all study sites using a centralized IVRS. The randomisation was stratified by site." Comment: Allocation concealment was adequate. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "To maintain concealment of the study‐group assignments, each study center used separate examining and treating neurologists (all of whom remained unaware of the assignments throughout the trial). Patients were instructed to take the assigned study drug at least 4 hours before study visits, in case patients in the BG‐12 groups had a side effect of flushing." Comment: The participants and personnel in this study were blinded to the allocated interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "To maintain concealment of the study‐group assignments, each study center used separate examining and treating neurologists (all of whom remained unaware of the assignments throughout the trial)." Comment: the outcomes assessors in this study were blinded to the allocated interventions |
| Incomplete outcome data (attrition bias) All outcomes | High risk | According to the Figure S2 (Patient Disposition) shown in Supplementary Appendix, 96 (23.1%) patients in high‐dose group, 95 (23.1%) patients in low‐dose group and 91 (22.2%) patients in placebo group withdrew from study. Comment: To a large degree, missing data and reasons were not balanced between groups. The high overall dropout rate of 22.8% over a period of 2 years had potential impacts on the results. Generally, the higher the ratio of participants with missing data to participants with events, the greater potential there is for bias, especially for the high frequency of events. The potential impact of missing continuous outcomes increases with the proportion of participants with missing data. Both MS relapse and the occurrence of adverse events are common events, a high risk of attrition bias existed. |
| Selective reporting (reporting bias) | Low risk | All listed outcomes were reported adequately. The risk of reporting bias was low. |
| Other bias | Unclear risk | Quote: "Members of the advisory committee and the data and safety monitoring committee collaborated with the sponsor, Biogen Idec, to develop the protocol and monitor the ongoing study. Data were collected by the investigators and were analysed by the sponsor." Comment: Conflicts of interest may exist. |
EDSS: Expanded Disability Status Scale MRI: magnetic resonance imaging MS: multiple sclerosis RRMS: relapsing‐remitting multiple sclerosis
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Kappos 2008 | RCT (NCT00168701) with a length of follow‐up shorter than one year |
| Schimrigk 2006 | This study was an uncontrolled pilot study |
RCT: randomised controlled trial
Differences between protocol and review
In the current review, we rephrased "disability progression" using "disability worsening". Only the primary benefit outcomes, the proportion of patients who discontinued study drug because of AEs and the proportion of patients with lymphopenia for low‐dose dimethyl fumarate administration were selected to create the 'Summary of findings' table because these outcomes are the most relevant to clinical practice. The restriction of "double‐blind studies" pre‐set in the protocol was removed. This change did not influence the conclusion of the review because all the trials eligible for the revised criteria were double‐blind. In addition, we removed duration of follow‐up and administration frequency as covariates for subgroup analysis in future updates.
Contributions of authors
All correspondence: Dian He Drafting of review versions: Zhu Xu and Dian He Search for trials: Feng Zhang and FangLi Sun Obtaining copies of trial reports: KeFeng Gu and Shuai Dong Selection of trials for inclusion/exclusion: Zhu Xu and Dian He Extraction of data: Zhu Xu and Dian He Entry of data: Zhu Xu and Dian He Interpretation of data analyses: Dian He
Sources of support
Internal sources
Guiyang Medical College, China.
External sources
The Science and Technology Department of Guizhou Province [No. LH(2014)7134], China.
Declarations of interest
HD ‐ none
XZ ‐ none
ZF ‐ none
SFL ‐ none
GKF‐ none
DS ‐ none
New
References
References to studies included in this review
Fox 2012 {published data only}
- Fox R, Miller D, Phillips JT, Kita M, Hutchinson M, Havrdova E, et al. Clinical efficacy of BG‐12 in relapsing‐remitting multiple sclerosis (RRMS): data from the phase 3 CONFIRM study. Meeting abstracts of the The American Academy of Neurology's 64th AAN Annual Meeting 2012.
- Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, et al. Placebo‐controlled phase 3 study of oral BG‐12 or glatiramer in multiple sclerosis. New England Journal of Medicine 2012;367(12):1087‐97. [DOI] [PubMed] [Google Scholar]
- Havrdova E, Miller D, Fox RJ, Phillips JT, Kita M, Hutchinson M, et al. Clinical and neuroimaging outcomes with BG‐12 treatment in CONFIRM (comparator and an oral fumarate in relapsing‐remitting multiple sclerosis) a multicenter, randomized, placebo‐controlled, phase‐3 study. European Journal of Neurology 2012;19:87. [Google Scholar]
- Havrdova E, Miller D, Fox RJ, Phillips JT, Kita M, Hutchinson M, et al. Clinical outcomes with BG‐12 treatment in CONFIRM (Comparator and an oral fumarate in relapsing‐ remitting multiple sclerosis) a multicentre, randomised, placebo‐controlled, phase 3 study. Journal of Neurology 2012;259(1 Suppl):36. [Google Scholar]
- Hutchinson M, Fox RJ, Miller D, Phillips JT, Kita M, Havrdova E, et al. Effect of BG‐12 in subgroups of patients with relapsing‐remitting multiple sclerosis: findings from the CONFIRM (comparator and an oral fumarate in relapsing‐remitting multiple sclerosis) study. European Journal of Neurology 2012;19:355. [Google Scholar]
- Hutchinson M, Fox RJ, Miller DH, Phillips JT, Kita M, Havrdova E, et al. Clinical efficacy of BG‐12 (dimethyl fumarate) in patients with relapsing‐remitting multiple sclerosis: subgroup analyses of the CONFIRM study. Journal of Neurology 2013;260(9):2286‐96. [DOI] [PubMed] [Google Scholar]
- Hutchinson M, Fox RJ, Phillips JT, Miller DH, Havrdova E, Kita M, et al. Efficacy and safety of BG‐12 (dimethyl fumarate) in relapsing‐remitting multiple sclerosis in the phase 3 CONFIRM study. Multiple Sclerosis 2013;19(5):683. [Google Scholar]
- Kita M, Fox RJ, Phillips JT, Hutchinson M, Havrdova E, Sarda SP, et al. Effects of BG‐12 (dimethyl fumarate) on health‐related quality of life in patients with relapsing‐remitting multiple sclerosis: findings from the CONFIRM study. Multiple Sclerosis 2014;20(2):253‐7. [DOI] [PubMed] [Google Scholar]
- Kita M, Fox RJ, Phillips JT, Hutchinson M, Havrdova E, Sarda SP, et al. Effects of BG‐12 on quality of life in patients with relapsing‐remitting multiple sclerosis: findings from the CONFIRM study. Multiple Sclerosis 2012;18(4):254. [DOI] [PubMed] [Google Scholar]
- Miller D, Fox R, Phillips JT, Kita M, Hutchinson M, Havrdova E, et al. Effects of BG‐12 on magnetic resonance imaging (MRI) endpoints in patients with relapsing‐remitting multiple sclerosis (RRMS): Data from the phase 3 CONFIRM study. Meeting abstracts of the The American Academy of Neurology's 64th AAN Annual Meeting 2012.
- Miller D, Fox RJ, Phillips JT, Kita M, Hutchinson M, Havrdova E, et al. Effects of BG‐12 on magnetic resonance imaging outcomes in CONFIRM (comparator and an oral fumarate in relapsing‐remitting multiple sclerosis), a randomized, placebo‐controlled, phase 3 study. Journal of Neurology 2012;259(1 Suppl):36‐7. [Google Scholar]
- Miller DH, Hutchinson M, Fox RJ, Phillips JT, Kita M, Havrdova E, et al. Effect of BG‐12 on magnetic resonance imaging activity in subgroups of patients with relapsing‐remitting multiple sclerosis: findings from the CONFIRM study. Multiple Sclerosis 2012;18(4):189‐90. [Google Scholar]
- Phillips JT, Fox R, Miller D, Kita M, Hutchinson M, Havrdova E, et al. Safety and tolerability of BG‐12 in patients with relapsing‐remitting multiple sclerosis (RRMS): analyses from the CONFIRM study. Meeting abstracts of the The American Academy of Neurology's 64th AAN Annual Meeting 2012.
Gold 2012 {published data only}
- Agarwal S, Kappos L, Gold R, Arnold D, Bar‐Or A, Giovannoni G, et al. Effects of BG‐12 on quality of life in patients with relapsing‐remitting multiple sclerosis: findings from the DEFINE study. Meeting abstracts of the The American Academy of Neurology's 64th AAN Annual Meeting 2012.
- Arnold D, Gold R, Bar‐Or A, Kappos L, Giovannoni G, Selmaj K, et al. Efficacy on MRI endpoints of BG‐12, an oral therapy, in relapsing‐remitting multiple sclerosis: data from the phase 3 DEFINE trial. Multiple Sclerosis 2011;17(10 Suppl):369‐70. [Google Scholar]
- Arnold D, Gold R, Kappos L, Bar‐Or A, Giovannoni G, Selmaj K, et al. Effect of BG‐12 on brain atrophy and lesions volume: MRI results from the define study during first and second year of treatment. Meeting abstracts of the The American Academy of Neurology's 64th AAN Annual Meeting.
- Arnold D, Gold R, Kappos L, Bar‐Or A, Giovannoni G, Selmaj K, et al. Effects of BG‐12 on magnetization transfer ratio in whole brain and normal‐appearing brain tissue: findings from the DEFINE study. Meeting abstracts of the The American Academy of Neurology's 64th AAN Annual Meeting 2012.
- Bar‐Or A, Gold R, Kappos L, Arnold DL, Giovannoni G, Selmaj K, et al. Clinical efficacy of BG‐12 (dimethyl fumarate) in patients with relapsing‐remitting multiple sclerosis: subgroup analyses of the DEFINE study. Journal of Neurology 2013;260(9):2297‐305. [DOI] [PubMed] [Google Scholar]
- Bar‐Or A, Gold R, Kappos L, Arnold DL, Giovannoni G, Selmaj K, et al. Effect of BG‐12 on magnetic resonance imaging activity in subgroups of patients with relapsing‐remitting multiple sclerosis: findings from the DEFINE study. European Journal of Neurology 2012;19:356. [Google Scholar]
- Giovannoni G, Gold R, Kappos L, Arnold D, Bar‐Or A, Selmaj K, et al. BG‐12 increases the proportion of patients free of clinical and radiologic disease activity in relapsing‐remitting multiple sclerosis: findings from the define study. Meeting abstracts of the The American Academy of Neurology's 64th AAN Annual Meeting 2012.
- Giovannoni G, Gold R, Kappos L, Arnold DL, Bar‐Or A, Selmaj K, et al. Analysis of clinical and radiological disease activity‐free status in patients with relapsing‐remitting multiple sclerosis treated with BG‐12: findings from the DEFINE study. Journal of Neurology 2012;259(1 Suppl):106. [Google Scholar]
- Gold R, Kappos L, Arnold DL, Bar‐Or A, Giovannoni G, Selmaj K, et al. Placebo‐controlled phase 3 study of oral BG‐12 for relapsing multiple sclerosis. New England Journal of Medicine 2012;367(12):1098‐107. [DOI] [PubMed] [Google Scholar]
- Kappos L, Gold R, Arnold D, Bar‐Or A, Giovannoni G, Selmaj K, et al. BG‐12 effects on patient‐reported outcomes in relapsing remitting multiple sclerosis: results from the DEFINE study. Multiple Sclerosis 2011;17(10 Suppl):488‐9. [Google Scholar]
- Kappos L, Gold R, Arnold DL, Bar‐Or A, Giovannoni G, Selmaj K, et al. Quality of life outcomes with BG‐12 (dimethyl fumarate) in patients with relapsing‐remitting multiple sclerosis: the DEFINE study. Multiple Sclerosis 2014;20(2):243‐52. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Kappos 2008 {published data only}
- Kappos L, Gold R, Miller DH, MacManus DG, Havrdova E, Limmroth V, et al. Effect of BG‐12 on contrast‐enhanced lesions in patients with relapsing‐remitting multiple sclerosis: subgroup analyses from the phase 2b study. Multiple Sclerosis 2012;18(3):314‐21. [DOI] [PubMed] [Google Scholar]
- Kappos L, Gold R, Miller DH, Macmanus DG, Havrdova E, Limmroth V, et al. Efficacy and safety of oral fumarate in patients with relapsing‐remitting multiple sclerosis: a multicentre, randomised, double‐blind, placebo‐controlled phase IIb study. Lancet 2008;372(9648):1463‐72. [DOI] [PubMed] [Google Scholar]
- MacManus DG, Miller DH, Kappos L, Gold R, Havrdova E, Limmroth V, et al. BG‐12 reduces evolution of new enhancing lesions to T1‐hypointense lesions in patients with multiple sclerosis. Journal of Neurology 2011;258(3):449‐56. [DOI] [PubMed] [Google Scholar]
Schimrigk 2006 {published data only}
- Schimrigk S, Brune N, Hellwig K, Lukas C, Bellenberg B, Rieks M, et al. Oral fumaric acid esters for the treatment of active multiple sclerosis: an open‐label, baseline‐controlled pilot study. European Journal of Neurology 2006;13(6):604‐10. [DOI] [PubMed] [Google Scholar]
Additional references
Albrecht 2012
- Albrecht P, Bouchachia I, Goebels N, Henke N, Hofstetter HH, Issberner A, et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. Journal of Neuroinflammation 2012;9:163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cella 1996
- Cella DF, Dineen K, Arnason B, Reder A, Webster KA, Karabatsos G, et al. Validation of the functional assessment of multiple sclerosis quality of life instrument. Neurology 1996;47(1):129‐39. [DOI] [PubMed] [Google Scholar]
Confavreux 2006
- Confavreux C, Vukusic S. Natural history of multiple sclerosis: a unifying concept. Brain 2006;129(3):606‐16. [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7(3):177‐88. [DOI] [PubMed] [Google Scholar]
Ermis 2013
- Ermis U, Weis J, Schulz JB. PML in a patient treated with fumaric acid. New England Journal of Medicine 2013;368(17):1657‐8. [DOI] [PubMed] [Google Scholar]
Filippini 2013
- Filippini G, Giovane C, Vacchi L, D'Amico R, Pietrantonj C, Beecher D, et al. Immunomodulators and immunosuppressants for multiple sclerosis: a network meta‐analysis. Cochrane Database of Systematic Reviews 2013, Issue 6. [DOI: 10.1002/14651858.CD008933] [DOI] [PubMed] [Google Scholar]
Fischer 1999
- Fischer JS, Rocca NG, Miller DM, Ritvo PG, Andrews H, Paty D. Recent developments in the assessment of quality of life in multiple sclerosis (MS). Multiple Sclerosis 1999;5(4):251‐9. [DOI] [PubMed] [Google Scholar]
Ghoreschi 2011
- Ghoreschi K, Brück J, Kellerer C, Deng C, Peng H, Rothfuss O, et al. Fumarates improve psoriasis and multiple sclerosis by inducing type II dendritic cells. Journal of Experimental Medicine 2011;208(11):2291‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gilgun‐Sherki 2004
- Gilgun‐Sherki Y, Melamed E, Offen D. The role of oxidative stress in the pathogenesis of multiple sclerosis: the need for effective antioxidant therapy. Journal of Neurology 2004;251(3):261‐8. [DOI] [PubMed] [Google Scholar]
GRADEpro 2008 [Computer program]
- Brozek J, Oxman A, Schünemann H. GRADEpro. Version 3.2 for Windows. Brozek J, Oxman A, Schünemann H, 2008.
Greenland 1985
- Greenland S, Robins JM. Estimation of a common effect parameter from sparse follow‐up data. Biometrics 1985;41(1):55‐68. [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. www.cochrane‐handbook.org.
Hutchinson 2014
- Hutchinson M, Fox RJ, Havrdova E, Kurukulasuriya NC, Sarda SP, Agarwal S, et al. Efficacy and safety of BG‐12 (dimethyl fumarate) and other disease‐modifying therapies for the treatment of relapsing‐remitting multiple sclerosis: a systematic review and mixed treatment comparison. Current Medical Research Opinion 2014; Vol. 30, issue 4:613‐27. [DOI] [PubMed]
Johnson 2010
- Johnson DA, Amirahmadi S, Ward C, Fabry Z, Johnson JA. The absence of the pro‐antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicological Sciences 2010;114(2):237‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Karampampa 2012a
- Karampampa K, Gustavsson A, Miltenburger C, Eckert B. Treatment experience, burden and unmet needs (TRIBUNE) in MS study: results from five European countries. Multiple Sclerosis 2012;18(2 Suppl):7‐15. [DOI] [PubMed] [Google Scholar]
Karampampa 2012c
- Karampampa K, Gustavsson A, Miltenburger C, Kindundu CM, Selchen DH. Treatment experience, burden, and unmet needs (TRIBUNE) in multiple sclerosis: the costs and utilities of MS patients in Canada. Journal of Population Therapeutics and Clinical Pharmacology 2012;19(1):11‐25. [PubMed] [Google Scholar]
Karampampa 2013b
- Karampampa K, Gustavsson A, Munster ET, Hupperts RM, Sanders EA, Mostert J, et al. Treatment experience, burden, and unmet needs (TRIBUNE) in Multiple Sclerosis study: the costs and utilities of MS patients in The Netherlands. Journal of Medical Economics 2013;16(7):939‐50. [DOI] [PubMed] [Google Scholar]
Kawalec 2014
- Kawalec P, Mikrut A, Wiśniewska N, Pilc A. The effectiveness of dimethyl fumarate monotherapy in the treatment of relapsing‐remitting multiple sclerosis: a systematic review and meta‐analysis. Current Neuropharmacology 2014;12(3):256‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kurtzke 1983
- Kurtzke JF. Rating neurological impairment in multiple sclerosis: an Expanded Disability Status Scale (EDSS). Neurology 1983;33:1444‐52. [DOI] [PubMed] [Google Scholar]
Lee 2008
- Lee DH, Linker RA, Gold R. Spotlight on fumarates. International MS Journal 2008;15(1):12‐8. [PubMed] [Google Scholar]
Lee 2012
- Lee DH, Gold R, Linker RA. Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: therapeutic modulation via fumaric acid esters. International Journal of Molecular Sciences 2012;13(9):11783‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Linker 2011
- Linker RA, Lee DH, Ryan S, Dam AM, Conrad R, Bista P, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011;134(3):678‐92. [DOI] [PubMed] [Google Scholar]
Litjens 2004
- Litjens NH, Burggraaf J, Strijen E, Gulpen C, Mattie H, Schoemaker RC, et al. Pharmacokinetics of oral fumarates in healthy subjects. British Journal of Clinical Pharmacology 2004;58(4):429‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lublin 1996
- Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 1996;46:907‐11. [DOI] [PubMed] [Google Scholar]
Lukashev 2007
- Lukashev M, Zeng W, Goelz S, Lee D, Linker R, Gold R, et al. Activation of Nrf2 and modulation of disease progression in EAE models by BG00012 (dimethyl fumarate) suggests a novel mechanism of action combining anti‐inflammatory and neuroprotective modalities. Multiple Sclerosis 2007;13(Suppl 2):149. [Google Scholar]
Mantel 1959
- Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. Journal of the National Cancer Institute 1959;22(4):719‐48. [PubMed] [Google Scholar]
McDonald 2001
- McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Annals of Neurology 2001;50(1):121‐7. [DOI] [PubMed] [Google Scholar]
National MS Society 2013
- National MS Society. Tecfidera. http://www.nationalmssociety.org/Treating‐MS/Medications/Tecfidera™ (accessed 2 September 2014).
Oleen‐Burkey 2012
- Oleen‐Burkey M, Castelli‐Haley J, Lage MJ, Johnson KP. Burden of a multiple sclerosis relapse: the patient's perspective. Patient 2012;5(1):57‐69. [DOI] [PubMed] [Google Scholar]
Peng 2012
- Peng H, Guerau‐de‐Arellano M, Mehta VB, Yang Y, Huss DJ, Papenfuss TL, et al. Dimethyl fumarate inhibits dendritic cell maturation via nuclear factor κB (NF‐κB) and extracellular signal‐regulated kinase 1 and 2 (ERK1/2) and mitogen stress‐activated kinase 1 (MSK1) signaling. Journal of Biological Chemistry 2012;287(33):28017‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Polman 2005
- Polman CH, Reingold SC, Edan G, Filippi M, Hartung HP, Kappos L, et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the "McDonald Criteria". Annals of Neurology 2005;58(6):840‐6. [DOI] [PubMed] [Google Scholar]
Polman 2011
- Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Annals of Neurology 2011;69(2):292‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Poser 1983
- Poser CM, Paty DW, Scheinberg L, McDonald WI, Davis FA, Ebers GC, et al. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Annals of Neurology 1983;13(8):227‐31. [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Scannevin 2012
- Scannevin RH, Chollate S, Jung MY, Shackett M, Patel H, Bista P, et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid‐derived 2)‐like 2 pathway. Journal of Pharmacology and Experimental Therapeutics 2012;341(1):274‐84. [DOI] [PubMed] [Google Scholar]
Schilling 2006
- Schilling S, Goelz S, Linker R, Luehder F, Gold R. Fumaric acid esters are effective in chronic experimental autoimmune encephalomyelitis and suppress macrophage infiltration. Clinical and Experimental Immunology 2006;145(1):101‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shirani 2012
- Shirani A, Zhao Y, Karim ME, Evans C, Kingwell E, Kop ML, et al. Association between use of interferon beta and progression of disability in patients with relapsing‐remitting multiple sclerosis. JAMA 2012;308(3):247‐56. [DOI] [PubMed] [Google Scholar]
Sweetser 2013
- Sweetser MT, Dawson KT, Bozic C. Manufacturer's response to case reports of PML. New England Journal of Medicine 2013;368(17):1659‐61. [DOI] [PubMed] [Google Scholar]
Tullman 2004
- Tullman MJ, Oshinsky RJ, Lublin FD, Cutter GR. Clinical characteristics of progressive relapsing multiple sclerosis. Multiple Sclerosis 2004;10(4):451‐4. [DOI] [PubMed] [Google Scholar]
U.S. Food and Drug Administration 2014
- U.S. Food, Drug Administration. FDA Drug Safety Communication: FDA warns about case of rare brain infection PML with MS drug Tecfidera (dimethyl fumarate). http://www.fda.gov/Drugs/DrugSafety/ucm424625.htm (accessed 25 November 2014).
van Oosten 2013
- Oosten BW, Killestein J, Barkhof F, Polman CH, Wattjes MP. PML in a patient treated with dimethyl fumarate from a compounding pharmacy. New England Journal of Medicine 2013;368(17):1658‐9. [DOI] [PubMed] [Google Scholar]
Vickrey 1995
- Vickrey BG, Hays RD, Harooni R, Myers LW, Ellison GW. A health‐related quality of life measure for multiple sclerosis. Quality of Life Research 1995;4(3):187‐206. [DOI] [PubMed] [Google Scholar]
Ware 1992
- Ware JE Jr, Sherbourne CD. The MOS 36‐item short‐form health survey (SF‐36). I. Conceptual framework and item selection. Medical Care 1992;30(6):473‐83. [PubMed] [Google Scholar]
Wilms 2010
- Wilms H, Sievers J, Rickert U, Rostami‐Yazdi M, Mrowietz U, Lucius R. Dimethyl fumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL‐1beta, TNF‐alpha and IL‐6 in an in‐vitro model of brain inflammation. Journal of Neuroinflammation 2010;7:30. [DOI] [PMC free article] [PubMed] [Google Scholar]