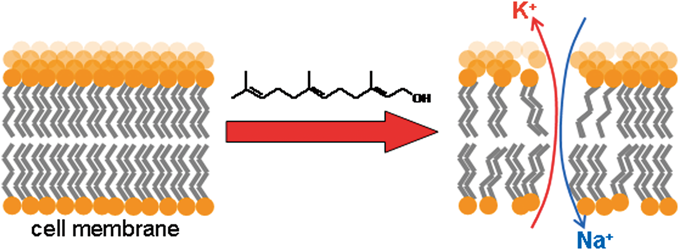
Aprepitant is a known inducer of CYP2C9, the main warfarin-metabolizing enzyme. Consequently, co-administration of these two drugs may result in reduction of the anticoagulation activity of warfarin. However, the nature and degree of time-dependent changes in prothrombin time international normalized ratio (PT-INR) after aprepitant and warfarin co-treatment in patients receiving anticancer chemotherapy has not been elucidated. We retrospectively examined the changes in warfarin dose, PT-INR, and warfarin sensitivity index (WSI; average of PT-INR value/average of daily warfarin dose) during four weeks,
i.e., one week before and three weeks after aprepitant administration. The mean and standard deviation values of WSI for one week before and one, two, and three weeks after the beginning of aprepitant administration were 0.51±0.22 (1.00,
n=34), 0.74±0.30 (1.53±0.59,
n=30), 0.38±0.15 (0.82±0.22,
n=28), and 0.46±0.29 (0.87±0.23,
n=24), respectively. Values in parentheses represent relative changes
versus WSI of one week before and number of subjects. Although the mean value of WSI significantly increased one week after aprepitant administration compared to that at one week before the administration, it in turn significantly decreased two weeks after compared to one week before (paired
t-test,
p<0.05 after Bonferoni correction). In patients taking warfarin, PT-INR should be carefully monitored for at least two weeks after the beginning of aprepitant administration because it may fluctuate with both aprepitant and chemotherapy during this period.
View full abstract