Practice Essentials
IgA vasculitis (IgAV)—previously known as Henoch-Schönlein purpura, although the eponymic name remains widely used—is a systemic inflammation of small vessels caused by an acute perivascular deposition of immunoglobulin A (IgA) and activation of neutrophils. [1] IgAV is classically characterized by the combination of cutaneous vasculitis (see the image below), arthritis, gastrointestinal (GI) tract, and kidney involvement, which can occur in flares. Rarely, the lungs or central nervous system (CNS) may be affected as well. [2] Variants of IgA may have involvement limited to skin or to the kidneys.
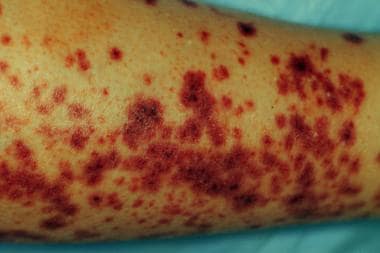
IgAV is the most common vasculitis in childhood. The incidence decreases with age, but in adults the severity increases with age. [3, 4, 5, 6, 7] Acute involvement of the GI tract impacts the short-term prognosis of the disease, while long-term prognosis depends on the severity of the kidney disease. Progression to chronic kidney failure may occur more than a decade after the onset of IgAV. [8, 9, 4, 10]
Signs and symptoms
The typical prodrome of IgAV includes the following:
-
Headache
-
Anorexia
-
Fever
Signs and symptoms usually develop insidiousliy, over days to weeks. The following are the most common:
-
Rash (95-100% of cases), especially involving the legs; this is the hallmark of the disease.
-
Joint pain (50-75% of children), especially involving the knees and ankles
-
Abdominal pain and vomiting
-
Subcutaneous edema
-
Scrotal edema
-
Bloody stools
-
Lower limb edema and hypertension (more common in adults) [7]
Because IgAV can affect all organ systems, a full physical examination is indicated.
Skin findings
Skin findings are usually the first sign of IgAV, preceding other symptoms by a mean of four days). [11, 7, 12] The rash begins with erythematous, macular, or urticarial lesions and progresses to blanching papules and later to palpable purpura; it is typically symmetrical and tends to be distributed in dependent body areas, such as the ankles and lower legs in older children and adults; the back and buttocks in toddlers; and the face, trunk, and upper extremities in young non-ambulant children. Facial involvement is a rare finding that is limited to more severe cases and never seen in isolation. [13] Hives, angioedema, and target lesions can also occur. The rash may be itchy but is rarely painful.
Localized subcutaneous edema is a common featur,e especially in children < 3 years old, usually occurring in dependent and periorbital areas. In adults it usually involves the dorsum of the hands. Blistering eruptions may also occur. [14]
Sometimes the rash may have a less typical presentation, including targetoid lesions. In adults, cutaneous involvement may be more severe, presenting as haemorrhagic blisters or necrotic skin lesions and often more extensive than in children. There is a variant of skin-limited IgAV called acute haemorrhagic oedema of infancy (AHEI) that rarely occurs after puberty.
Joint findings
Arthritis/arthralgia occurs in 74% of patients and is the presenting feature in as many as 15% of pediatric cases. Joints may be swollen, tender, and painful; however, warmth, erythema, and effusions are not typical. The knees and ankles are most commonly affected. Young children with lower extremity involvement will refuse to walk. Rarely, the fingers and wrists may be involved. The arthritis is usually transient or migratory and oligoarticular (one to four joints involved). Joint involvement may precede the onset of purpura but usually not by more than one or two days. [11]
Kidney findings
Kidney involvement may occur in 40-50% of cases, especially in older children and adults, and is the leading cause of morbidity in IgAV, although it may be asymptomatic. The most common manifestation of kidney involvement is microscopic hematuria, followed by proteinuria without edema. Gross hematuria may occur in the acute phase of the disease but is uncommon and usually short-lived. IgAV can present as acute nephritis or nephritis associated with nephrotic syndrome [13]
Gastrointestinal findings
GI findings may precede skin manifestations by a few days to a week. Approximately 50% of children with IgAV have GI manifestations. These range from mild (nausea, vomiting, and cramping abdominal pain) to severe (severe abdominal pain, hematemesis or melena, bloody diarrhea, massive GI hemorrhage). [15]
Other findings
Vasculitis involving the following has been reported in IgAV:
-
Myocardium
-
Lungs
-
Genitals (scrotal edema, orchitis, penile edema, priapism in males)
-
Central nervous system (CNS) – Intracranial hemorrhage with confusion, convulsions, weakness, visual changes, and reduced level of consciousness [16]
-
Adrenal gland
-
Pancreas [17]
See Presentation for more detail.
Diagnosis
IgAV is a clinical diagnosis, although confirmation by histologic analysis from skin or kidney biopsy is sometimes helpful. The diagnosis of IgAV, as revised by the European League Against Rheumatism (EULAR), Paediatric Rheumatology International Trial Organisation (PRINTO), and Paediatric Rheumatology European Society (PRES), requires the presence of palpable purpura or petechiae—with lower limb predominance and without thrombocytopenia or coagulopathy; usually in clusters—along with at least one of the following [18, 19, 20] :
-
Diffuse abdominal pain of acute onset
-
Any biopsy sample showing leukocytoclastic vasculitis, or proliferative glomerulonephritis with predominant IgA deposition in a kidney biopsy sample in patients with atypical distribution of purpura
-
Acute arthritis or acute arthralgia in any joint
-
Kidney involvement (any hematuria or proteinuria) and IgA deposition
No specific diagnostic laboratory test is available to assess for markers of IgAV. The following general laboratory tests may be helpful for excluding other diagnoses and evaluating kidney function:
-
Antinuclear antibody (ANA) and rheumatoid factor (RF)
-
Factors VIII and XIII
-
Urinalysis (may show dysmorphic red blood cells and casts with an elevated protein:creatinine ratio)
-
Complete blood count (CBC)
-
Platelet count
-
Erythrocyte sedimentation rate (ESR)
-
Stool guaiac test
-
Blood urea nitrogen (BUN) and creatinine
-
Amylase and lipase
-
Electrolytes
-
Plasma D-dimer
-
Plasma thrombin-antithrombin (TAT) complex, prothrombin fragment (PF)-1, and PF-2
-
Prothrombin time (PT) and activated partial thromboplastin time (aPTT)
-
Serum IgA (elevated in 50-70% of patients with IgAV; higher levels are associated with kidney involvement)
-
Antistreptolysin O (ASO)
-
CH50
-
C3 and C4
-
Immunocomplexes of IgG and IgA
Imaging modalities that may be considered include the following:
-
Ultrasonography (abdominal, scrotal/testicular)
-
Radiography (chest radiography, plain radiography of the abdomen, contrast radiography of the small intestine, barium enema study)
-
Magnetic resonance imaging (MRI; for assessing neurologic findings)
-
Computed tomography (CT) of the head or abdomen
Other studies that may be warranted are as follows:
-
Endoscopy - Purpuric lesions may be seen in the descending duodenum, stomach, and colon. The terminal ileum and jejunum may also be involved, with submucosal edema, ulceration, and spasm.
-
Kidney biopsy (particularly when nephrotic syndrome persists and when kidney function deteriorates). The severity of kidney involvement correlates with findings on kidney biopsy, Light microscopic findings can range from mild mesangial proliferation to crescentic glomerulonephritis. Diffusion mesangial IgA deposits with co-deposition of C3 complement (seen in about 75% of cases) is the hallmark of the disease. The absence of C1q and C4 classical complement pathway components distinguishes glomerulonephritis from other forms of immune-mediated glomerulonephritis (eg, lupus nephritis).
See Workup for more detail.
Management
Treatment remains primarily supportive in most cases, though pharmacotherapy, plasmapheresis, and surgical interventions may also be considered in select cases.
Supportive measures may include the following:
-
Ensuring adequate hydration
-
Monitoring for GI and kidney complications
-
Treating minor symptoms of arthritis, edema, fever, or malaise
-
Eating a bland diet
-
Discontinuing any drugs suspected of playing a causative role
Joint and soft tissue discomfort may be reduced by giving analgesics, such as the following:
-
Acetaminophen
-
Ibuprofen
-
Flurbiprofen
-
Ketoprofen
-
Naproxen
Corticosteroids may be considered in the following situations:
-
Persistent nephrotic syndrome
-
Crescents in more than 50% of glomeruli
-
Severe abdominal pain
-
Substantial GI hemorrhage
-
Severe soft tissue edema
-
Severe scrotal edema
-
Neurologic system involvement
-
Intrapulmonary hemorrhage
The beneficial effect of methylprednisolone pulses has been shown in patients receiving combinations of multiple immunosuppressive drugs.
In IgAV nephritis, a disease considered to be benign, long-term follow-up studies showed delayed development of chronic kidney disease in this population in the absence of rapidly progressive glomerulonephritis when steroids and other immunosuppressants were used.
Other treatment regimens have included IV or oral steroids with or without any of the following:
-
Azathioprine
-
Cyclophosphamide
-
Cyclosporine
-
Hydroxychloroquine
-
Angiotensin-converting enzyme inhibitors or angiotensin receptor blockers
-
Dipyridamole
-
High-dose IV immunoglobulin G (IVIg)
-
Danazol
Plasmapheresis may be effective in delaying the progression of kidney disease. Angiotensin converting enzyme inhibitors or receptor blockers may be given for moderately severe proteinuria.
Surgical interventions that may be considered in specific circumstances include the following:
-
Surgery for severe bowel ischemia
-
Kidney transplantation for severe kidney disease that is resistant to medical therapy
-
Tonsillectomy together with corticosteroid pulse therapy for progressive IgAV nephritis
See Treatment and Medication for more detail.
For patient education information, see Henoch-Schonlein Purpura.
Background
IgA vasculitis (IgAV) was first described in 1802 by the English physician William Heberden, in two boys who presented with abdominal pain, purpuric rash, and arthralgia. [21] The historical term for the condition honors the German pediatrician Eduard Heinrich Henoch and his teacher Johann Lukas Schönlein, who described the association of non-thrombocytopenic purpura with joint pain in 1837 and called the condition purpura rheumatica. [22] Henoch added GI and kidney involvement in 1874. [23] In addition to Henoch-Schönlein purpura, IgAV has also been referred to as Schönlein-Henoch purpura, anaphylactoid purpura.
IgAV is an acute IgA–mediated disorder characterized by a generalized vasculitis involving the small vessels of the skin, the gastrointestinal (GI) tract, the kidneys, the joints, and, rarely, the lungs and the central nervous system (CNS). [3, 24] It is a subset of necrotizing vasculitis characterized by fibrinoid destruction of blood vessels and leukocytoclasis.
The prevalence of IgAV peaks in children aged 3-10 years, with the mean age at presentation being 6 years, but the condition is also seen in adults. [25] It has been reported that IgAV will occur in 10-30 per 100,000 children younger than 17 years will develop IgAV. There is a strong ethnic influence on the incidence of the disease, with approximately 70 cases per 100,000 children annually in Asia. [26] In the Northern Hemisphere, the disease occurs mostly between November and January. IgAV has typically been reported to be more common in males, with a male-to-female ratio ranging from 1.5-2:1, but some studies have found a more equal distribution between the sexes. [27, 28]
The dominant clinical features of IgAV include cutaneous purpura, arthritis, abdominal pain, and nephritis. These manifestations may develop over the course of days to weeks and may vary in their order of presentation. The classic rash of IgAV is not the initial presenting sign in approximately one-quarter of patients. In one half to two thirds of children, an upper respiratory tract infection precedes the clinical onset of IgAV by 1-3 weeks. In general, patients with IgAV appear mildly ill. They often have a low-grade fever, with a temperature that usually does not exceed 38°C (100.4°F).
IgAV is typically an acute, self-limited illness, and treatment is primarily supportive. In the majority of children, symptoms and signs of IgAV resolve within several days or months and the outcome is excellent. However, one third of patients have 1 or more recurrences.
Kidney involvement is the most important determinant of long-term morbidity. Up to 30-50% of children present with hematuria and/or proteinuria, or develop it within 4-6 weeks of the initial presentation. This is usually mild and self-limiting. However, approximately 20% of IgAV children with nephritis (7% of all IgAV cases) will develop either a nephritis or nephrotic syndrome. [29, 30, 31, 32, 33, 34]
Keratitis and uveitis are rare sequelae of eye involvement.
Pathophysiology
IgA clearly plays a critical role in the immunopathogenesis of IgAV, as evidenced by increased serum IgA concentrations, IgA-containing circulating immune complexes, and IgA deposition in vessel walls of affected organs and in the kidney mesangium. Deposition of IgA aggregates or IgA complexes in target organs occurs with activation of the alternative complement pathway (with deposition of C3). This results in elaboration of inflammatory mediators, including vascular prostaglandins such as prostacyclin, that may play a central role in the pathogenesis of IgAV and its organ-specific clinical manifestations. [35]
IgA is found in the serum and mucosal secretions and is a major class of immunoglobulins that plays an important role in mucosal immunity. IgAV is almost exclusively associated with abnormalities involving IgA1, rather than IgA2. IgA1 is phylogenetically younger and differs from IgA2 by the insertion of a 13-17 amino acid sequence in the hinge region of the IgA1 molecule. [36] The predominance of IgA1 in IgAV may be a consequence of abnormal glycosylation of O-linked oligosaccharides unique to the IgA1 hinge region. Patients with IgAV and IgA nephritis express inherited galactose-deficient glycosylation of IgA1 molecules. [37]
IgAV-associated nephritis is characterized by an abnormal IgA1 glycosylation pattern with reduced galactosylation. [38] The hinge region of IgA1 contains up to six major glycosylation sites at serine and threonine residues. The O-glycans include a core N-acetylgalactosamine (GalNAc), which is usually extended with galactose to form Galβ1,3GalNAc, which can bind to N-acetylneuraminic acid (Neu5Ac). Thus, each IgA1 O-glycan can have one of four short carbohydrate structures (types III, IV, V, and VI), leading to a mixture of IgA1 forms with varying degrees of galactosylation.
Patients with IgAV-associated nephritis have a high prevalence of galactose-deficient (types I and II) IgA1. [38] The lack of terminal β1,3-galactosyl residues in the hinge region of IgA might be due to the reduced activity of β1,3-galactosyltransferase in IgA1-producing peripheral B cells. This reduction of galactosylation results in the exposure of GalNAc residues in the IgA1 surface, forming a novel antigen and inducing a humoral IgG autoimmune response. [39]
Circulating complexes of mixed IgG and galactose-deficient IgA1 are detected in patients with IgAV, and also in the serum of patients with mucosal infections. [40] The finding that galactose-deficient IgA1 molecules are found in IgAV only during an episode of nephritis lends support to the pathophysiologic role of galactose-deficient IgA1 molecules in IgAV nephritis. [41]
A subpopulation of human lymphocytes bears surface Fc and/or C3 receptors (complement receptor lymphocytes), which can bind circulating immune complexes or C3 generated by activation of the alternative complement pathway. Such immune complexes appear in IgAV and may be part of the pathogenetic mechanism.
Some have speculated that an antigen stimulates the production of IgA, which, in turn, causes vasculitis. Allergens, such as foods, horse serum, insect bites, exposure to cold, and drugs (eg, ampicillin, erythromycin, penicillin, quinidine, quinine, tumor necrosis factor alpha blockers), may precipitate the illness. [42]
Infectious triggers include bacteria (eg, Haemophilus parainfluenzae, Mycoplasma, Legionella, Yersinia, Shigella, Salmonella, Helicobacter pylori) and viruses (eg, adenoviruses, Epstein-Barr virus [EBV], parvovirus B19, varicella-zoster virus [VZV]). Vaccines such as those against measles, hepatitis B, influenza, typhoid, cholera, and yellow fever have also been implicated. [43] However, there is no evidence supporting a direct role of herpesvirus or retrovirus infection in the pathogenesis of IgAV. [35]
Alterations in the production of interleukins (ILs) and growth factors may also play a pathogenetic role. Tumor necrosis factor (TNF), IL-1, and IL-6 may mediate the inflammatory process present in IgAV. Transforming growth factor (TGF)–β is a recognized stimulant of IgA production. The elevated levels of hepatocyte growth factor present during the acute phase of IgAV may reflect endothelial-cell damage or dysfunction. Increased levels of vascular endothelial growth factor (VEGF) may at least partly induce these changes.
Cytokines have been implicated in the pathogenesis of IgAV, and endothelins (ETs), which are vasoconstrictor hormones produced by endothelial cells, may also have a role. Levels of ET-1 are substantially higher during the acute phase of the disease than during remission or in healthy children. However, ET-1 levels do not appear to correlate with morbidity, severity of disease, or acute-phase reactant response. [43]
Although several lines of evidence suggest a genetic susceptibility to IgAV, the fundamental basis for this abnormality remains unclear. A functional correlation of the IL1RN-2 allele and IL-1ra production in patients with IgA nephropathy and IgAV nephritis has been described. Therefore, gene polymorphism may contribute to the diversity of clinical responses to inflammatory stimulation. The prevalence of the human parvovirus B19 component NS1 gene in patients with IgAV and hypersensitivity vasculitis is increased.
Gershoni-Baruch et al showed that in the Israeli population, 10% of patients with IgAV were homozygous for mutations in MEFV (the gene defective in familial Mediterranean fever that encodes the protein pyrin/marenostrin, which regulates caspase-1 activation and IL-1b production). An additional 17% of patients with IgAV had heterozygous defects of this gene. [44] Peru et al reported an increased risk of IgAV in children carrying HLA A2, A11, and B35 antigens and a reduced risk in those carrying HLA A1, B49, and B50 antigens. [45]
Researchers are currently investigating the importance of nitric oxide (NO) production in disease activity. Inducible NO synthase polymorphism has been associated with susceptibility to IgAV in northwestern Spain. Aliyazicioglu et al have suggested that leptin and NO may play a role in the immunoinflammatory process of IgAV, especially in the acute phase. [46]
Yilmaz et al examined markers of hypercoagulability in 28 children with IgAV and 79 healthy children, and found that levels of fibrinogen, D-dimer, thrombin-antithrombin (TAT) complex, prothrombin fragment (PF)-1, PF-2, and von Willebrand factor antigen (vWAg) and its activity (RiCof) were significantly higher during the acute phase than during the recovery phase and were significantly higher in patients with IgAV than in control subjects. [47] The severity of disease correlated significantly with TAT, PF-1, PF-2, vWAg, and D-dimer levels. Higher levels of matrix metalloproteinase–9 (MMP-9) in urine and serum appear to correlate with increased nephrologic severity in children with IgAV.
IgA vasculitis versus IgA nephropathy
IgAV and IgA nephropathy appear to be related disorders. However, the precise relation between them requires further definition. The question has been raised as to whether IgAV and IgA nephropathy are two aspects of a single disease entity or two distinct entities. The following commonalities and differences have been noted:
-
In IgAV-associated nephritis, repeated and prolonged episodes of acute glomerular inflammation lead to fibrous scars and hyperfiltration in the remaining areas, resulting in chronic kidney disease that may progress to chronic kidney failure. Thus, the number and severity of acute episodes of IgAV-associated nephritis have a crucial role in the subsequent progression and loss of kidney function. In IgA nephropathy, initiation and progression to chronic kidney failure occurs slowly and often is asymptomatic, with the development of glomerulosclerosis and tubulointerstitial fibrosis. [51]
-
Extrarenal manifestations in IgA nephropathy are similar to those in IgAV.
-
IgA nephropathy has developed in patients with a history of IgAV, and IgAV and IgA nephropathy have occurred in the same families; in a French survey of 40 families in which two or three members had IgA nephropathy, five families had one or two members with IgAV. [52]
-
Patients with IgAV who undergo kidney transplantation develop IgA deposits in the graft.
-
The prevalence of both conditions is high in certain geographic areas.
-
Cystic changes in the ovaries of a prepubertal girl with IgAV have been recorded. [56]
Overall, the data support the view that IgAV and IgA nephropathy are distinct diseases. [57] Zhou et al examined 31 children aged 3-15 years with IgA nephropathy and 120 children aged 4-15 years with IgAV, noting their clinical manifestations, blood biochemistries, serum immunology, and follow-up data. [58] Pathologic findings on light microscopy, immunofluorescence study, and electron microscopy of kidney biopsy specimens were analyzed and compared between 31 children with IgA nephropathy and 32 children with IgAV.
The age of onset was older than 12 years in 25.8% of the children with IgA nephropathy but in only 10% of those with IgAV. [58] Clinical patterns of IgA nephropathy were similar to those of IgAV, but extrarenal manifestations were observed more often in patients with IgAV. All of the IgAV patients had purpura, 59% had GI symptoms, and 47% had arthralgia. Of the children with IgA nephropathy, only 3.2% had abdominal pain. [58] Kidney pathology in patients with IgA nephrology versus those with IgAV was as follows:
-
Global sclerosis: 35.5% vs 3.1%
-
Mesangial sclerosis: 41.9% vs 6.3%
-
Endothelial proliferation: 29% vs 65.6%
-
Thin basement-membrane nephropathy: 6.5% vs 0%
In the kidneys of patients with IgAV, electronically dense deposits were sparse, loose, and widely spread in the glomerular mesangium, subendothelial area, and intra-basement membranes. In those with IgA nephropathy, the deposits were dense, lumpy, and mostly limited to mesangium and paramesangium. [58]
Immunoglobulin G (IgG) was found in glomerular immune deposits in 71.9% of patients with IgAV but in only 19.4% of patients with IgA nephropathy. [58] No IgG deposit was observed in 81.6% of those with IgA nephropathy; most had IgA and immunoglobulin M (IgM) or C3 deposits. Predominant IgG deposits were found in 12.5% of IgAV patients, with relatively weak IgA deposits. Moreover, 6.3% of IgAV patients had linear IgG deposits in the glomerular capillary wall, a finding that was not noted in patients with IgA nephropathy.
In patients with IgAV, the complete remission rate was 72.5%, on follow-up at an average of 20 months; in those with IgA nephropathy, the corresponding rate was 19.4% on follow-up after 34 months. [58] Moreover, 64.5% of patients with IgA nephropathy had consistent hematuria and proteinuria, and 16.1% had active nephritides.
The important clinicopathologic differences Zhou et al found between IgAV and IgA nephropathy argue against the single-disease hypothesis.
Etiology
The etiology of IgAV remains to be clearly defined but is thought to be multifactorial, with genetic, environmental, and antigenic components. More than 75% of patients report antecedent upper respiratory tract or GI infection. Multiple bacterial and viral infectious agents have been associated with the development of IgAV, and cases also have been reported in association with medication use and vaccinations. [59, 60]
Infections that may precede the development of IgAV include the following:
-
Group A streptococcal infection (most common)
-
Infectious mononucleosis
-
Subacute bacterial endocarditis
-
Hepatitis
-
Mycoplasma infection
-
Campylobacter enteritis
-
Yersinia infection
-
Shigella infection
-
Salmonella infection
-
Legionellainfection
-
Parvovirus infection
-
Adenovirus infection
-
Epstein-Barr virus (EBV) infection
-
Varicella-zoster virus (VZV) infection
-
Rotavirus infection
Vaccinations that may precede the development of IgAV include the following [35, 43] :
-
Measles, mumps, rubella (MMR)
-
Hepatitis B
-
Influenza
-
Typhoid and paratyphoid A and B
Environmental exposure to the following may precede the development of IgAV:
-
Foods
-
Horse serum
-
Cold temperatures
-
Insect bites
Glomerulocystic kidney disease has also been noted as a risk factor.
Epidemiology
United States statistics
In the United States, the incidence of IgAV is approximately 14-15 cases per 100,000 population in children, compared with 1-3 cases per 100,000 per year in adults. [68, 69, 70, 71]
International statistics
Worldwide, the incidence rates of IgAV in children and adolescents aged < 15 years vary widely, ranging from 3.5 per 100,000 persons in Japan to 26.7 per 100,000 persons in Scotland. [72] In the United Kingdom overall, the estimated annual incidence of IgAV is 20.4 cases per 100,000 population aged < 17 years, with a peak incidence of 70 per 100,000 in children of age four to six years. [26] Incidence rates reported elsewhere in Europe are 17.5 per 100,000 in Sweden and 18.6 per 100,000 persons in France. [72] Surveys from Taiwan and the Czech Republic report a lower incidence of 10 per 100,000 in children < 17 years of age, with a peak incidence at five to seven years of age. [73, 74]
In a study that examined the kidney biopsy results of 65 children younger than 18 years obtained by the Clinical Hospital in the Croatian region of Dalmatia over 10 years (1995-2005), 10.8% of glomerulonephritis cases were due to IgAV. [75]
From January 1983 to June 2004, Suehiro et al followed 4502 patients at the Pediatric Rheumatology clinic in Brazil. [76] A diagnosis of IgAV was made in 203 cases (4.5%); 5 patients (0.1%) had acute hemorrhagic edema of infancy (AHEI). All patients with AHEI were male, and the mean age at onset was 18 months (range, 8-21 months).
Age-related demographics
IgAV primarily affects children; it may be seen in adults, but much less frequently. [24, 4] In the United States, the prevalence peaks in children aged 5 years. Approximately 75% of cases occur in children aged 2-11 years; IgAV is rare in infants and young children. Acute hemorrhagic edema of infancy (AHEI), a related but milder condition, occurs in infants younger than 2 years. [77] In adults, the mean age of onset of IgAV is 50 years. [78]
In a study of patients aged 14 years and older seen at a hospital in southern Spain, the incidence of IgAV was 1.5 per million population. [79] In a study of patients aged 17 years and older seen in a Norwegian community hospital, the prevalence of IgAV was 3.3 cases per 100,000 inhabitants. [80]
Older age at the onset of IgAV is associated with the development of chronic kidney disease. [12]
A retrospective study of children with IgAV treated at a hospital in Indonesia found that gastrointestinal manifestations tended to occur in patients younger than 5 years old, while kidney involvement tended to occur in those 11-15 years old. The study comprised 128 patients, ranging from 6 months to 15 years of age, from 2006 to 2011. Peak morbidity was between 5-10 years of age. In most patients (71%) purpura was the first symptom; 71 patients (44.5%) had arthritis and 89 patients (69.5%) had abdominal pain, while 28 patients (21.8%) had kidney involvement. [81]
A multi-institutional German study that included data from 202 pediatric patients with IgAV nephritis reported that children older than 10 years of age had more insidious onset of non-nephrotic proteinuria, impaired kidney function, longer delay to biopsy, and more chronic histopathologic lesions than younger children. [82]
Sex-related demographics
In children, IgAV has typically been reported to be more common in boys, with a male-to-female ratio ranging from 1.5-2:1, but some studies have found a more equal distribution between the sexes. [27, 28] In adults, the male-to-female ratio is approximately 1:1.
Race-related demographics
Whites are affected more often than Blacks.
In a study from Thailand, patients most commonly presented between the ages of 3 and 5 years. [83] Frequency peaked from December to February. Organs involved included the skin (100%), GI tract (74.5%), and kidneys (46.8%). Joints were also affected (42.6%). Kidney involvement was detected within the first 2 months in 16 patients (72.7%); however, it was delayed until 6 months after diagnosis in 6 patients. No risk factors for kidney involvement could be identified. On mean follow-up of 2.6 years (range, 1-5 years), residual kidney disease occurred in 6 (38%) of 16 patients, but none had chronic kidney failure.
In a study from China, a male predominance was observed in children but not in adults. [84] Preceding infection was noted in 40.5% of children and 31.6% of adults; 8.3% of children and 13.2% of adults were receiving medication at the onset of the disease. Abdominal pain was more common in children than adults (70.2% vs 28.9%), but kidney involvement was more common and severe in adults than in children; this involvement frequently manifested as hypertension and heavy proteinuria. [84] During acute attacks, leukocytosis, thrombocytosis, and elevated serum C-reactive protein (CRP) levels were most frequently observed in children, whereas elevated serum IgA and cryoglobulin levels were most common in adults.
A study of 450 cases from Turkey showed that girls, patients with atypical presentations, and patients undergoing early corticosteroid treatment had an increased risk of developing kidney disease; relapses occurred more often in children treated with corticosteroids. [85]
Familial kindreds with IgAV have been noted in Taiwanese aboriginal people. [86]
Prognosis
IgAV is generally a benign disease with an excellent prognosis. Spontaneous resolution is usual: Most patients experience complete resolution of symptoms within 8 weeks, and probably fewer than 5% experience chronic symptoms. However, initial attacks of IgAV can last several months, and relapses are possible. IgAV is fatal only in the rarest of cases.
A clinical course with complete resolution of the disease usually occurs in patients with the following:
-
Mild kidney involvement
-
No neurologic complications
-
Disease that lasts less than 4-6 weeks initially
Children younger than 3 years usually have a shorter, milder course than older patients do, as well as fewer recurrences.
Recurrences occur in as many as 50% of patients within 6 weeks but can happen as late as 7 years after the initial disease. A study by Calvo-Río et al indicated that in patients with IgAV, the chance of relapse is greater in those with GI and joint manifestations: the relapse rate in patients with abdominal pain was 72.3%, compared with 62.3% in those without abdominal pain; the relapse rate in patients with joint manifestations was 27.8%, compared with 15.5% in those without joint manifestations. [87]
Although IgAV generally resolves without permanent consequences, serious GI and renal complications may occur, and the higher the number of recurrences, the higher the likelihood of permanent kidney damage. Potential GI complications include the following:
-
Intussusception (usually ileoileal)
-
Bowel infarction
-
Bowel perforation
-
Hydrops of the gallbladder
-
Massive GI bleeding
Kidney damage related to IgAV is the primary cause of morbidity and mortality. As many as 15% of patients may have long-term kidney insufficiency, but no more than 1-2% will have ESKD. As many as 20% of the children with IgAV treated in specialized centers require hemodialysis. The risk of kidney damage appears to be worse in adults than in children (in particular, those aged ≤6 years).
Predictors of serious nephropathy or end-stage kidney disease (ESKD) include bloody stools and persistent rash. Initial kidney manifestations and outcomes are as follows [38] :
-
Nephrotic and nephritic syndrome - Progression to chronic kidney disease (CKD) in > 50% of cases
-
Nephrotic syndrome - Progression to CKD in ±40% of cases
-
Nephritic syndrome - Progression to CKD in ±15% of cases
-
Heavy non-nephrotic proteinuria - Progression to CKD in ±15% of cases
-
Hematuria and/minimal proteinuria - Progression to CKD in < 5% of cases
-
Nephrotic syndrome persisting for < 3 months - Almost no progression to ESKD
-
Nephrotic syndrome persisting for > 3 months - Progression to ESRD in ±40% of cases.
However, patients with a normal urinalysis at 6 months and without previous renal involvement have not gone on to develop kidney problems. [88]
Pregnant women who had IgAV during childhood appear to be at increased risk for developing hypertension and proteinuria during pregnancy. [89]
The long-term prognosis for patients with IgAV nephritis is determined by the development of CKD, which sometimes is difficult to predict from the initial clinical and histologic presentation. CKD can develop long-term even after apparent complete recovery from IgAV nephritis. [38]
Patient Education
Patients should be informed that the disease is most likely to resolve with few residual adverse effects but that relapses are possible. The clinician should explain that severe kidney involvement is rare but that aggressive treatment may be required if it does occur.
For patient education information, see Henoch-Schonlein Purpura and IgA Vasculitis.
-
Purpuric papules and plaques of the lower extremity characteristic of IgA vasculitis (Henoch-Schönlein purpura).
-
Hemorrhagic macules, papules, and patches on the ankle and foot of a child with IgA vasculitis (Henoch-Schönlein purpura).
-
Typical rash distribution of IgA vasculitis (Henoch-Schönlein purpura).
-
Characteristic rash of IgA vasculitis (Henoch-Schönlein purpura).
-
Older lesions of IgA vasculitis (Henoch-Schönlein purpura) demonstrating increased extravasation with ecchymoses on dorsal foot and ankle.
-
A 9-year-old boy with IgA vasculitis (Henoch-Schönlein purpura). Note confluence of purpura around the ankles. Image courtesy of Pamela L Dyne, MD.
-
A 7-year-old girl with IgA vasculitis (Henoch-Schönlein purpura). Image courtesy of Pamela L Dyne, MD.







