

Abstract
Intelligence in childhood, as measured by psychometric cognitive tests, is a strong predictor of many important life outcomes, including educational attainment, income, health and lifespan. Results from twin, family and adoption studies are consistent with general intelligence being highly heritable and genetically stable throughout the life course. No robustly associated genetic loci or variants for childhood intelligence have been reported. Here, we report the first genome-wide association study (GWAS) on childhood intelligence (age range 6–18 years) from 17 989 individuals in six discovery and three replication samples. Although no individual single-nucleotide polymorphisms (SNPs) were detected with genome-wide significance, we show that the aggregate effects of common SNPs explain 22–46% of phenotypic variation in childhood intelligence in the three largest cohorts (P = 3.9 × 10−15, 0.014 and 0.028). FNBP1L, previously reported to be the most significantly associated gene for adult intelligence, was also significantly associated with childhood intelligence (P = 0.003). Polygenic prediction analyses resulted in a significant correlation between predictor and outcome in all replication cohorts. The proportion of childhood intelligence explained by the predictor reached 1.2% (P = 6 × 10−5), 3.5% (P = 10−3) and 0.5% (P = 6 × 10−5) in three independent validation cohorts. Given the sample sizes, these genetic prediction results are consistent with expectations if the genetic architecture of childhood intelligence is like that of body mass index or height. Our study provides molecular support for the heritability and polygenic nature of childhood intelligence. Larger sample sizes will be required to detect individual variants with genome-wide significance.
Keywords: intelligence, IQ, cognitive, association, FNBP1L, polygenic
Introduction
Intelligence in human populations is associated with a wide range of important life outcomes, including educational attainment, income, health and longevity, and intelligence in childhood is a predictor of those outcomes.1 Twin, family and adoption studies have shown that intelligence, as measured using validated psychometric cognitive tests (Intelligence Quotient (IQ)-type tests), is one of the most heritable behavioural traits.2 These findings have been consistently replicated, but the molecular basis of intelligence remains poorly understood. The supporting evidence from the molecular findings has not been consistent and many reported candidate–gene associations have not been replicated.2 A recent study suggested that most reported associations between candidate genes and intelligence are likely to be false.3
The recent successes of genome-wide association studies (GWAS) for many complex traits, where > 1200 genetic variants have been associated with complex traits,4 have not been achieved for behavioural traits, including intelligence.5–8 The most plausible reason for this failure is that the effect size of individual genetic variants is so small that the current experimental sample sizes are not large enough for detection.9,10 For example, using ∼3500 individuals, Davies et al.6 did not find any genome-wide significant single-nucleotide polymorphism (SNPs) associated with intelligence in adults. However, when the combined effects of SNPs were analysed simultaneously, they found that common SNPs accounted for 40–51% of the variation for different measures of intelligence.
Whereas intelligence shows remarkable stability throughout the life course, age-to-age change also occurs. A correlation between intelligence score from age 11 to age 77 of 0.63 has been reported.11 It has also been consistently shown that heritability increases from childhood (h2 = 0.41) to young adulthood (h2 = 0.66).12 To date, several GWAS results on intelligence have been reported,5–8 but the only published GWAS results7,8 for childhood intelligence were based on DNA pooling, where SNP genotyping was carried out on pools made of DNA from many individuals.
Understanding individual differences in childhood intelligence can contribute to dissecting its observed association with important outcomes later in life. The aim of this study is to elucidate the genetic and environmental bases of childhood intelligence by identifying associated genetic variants and estimating their contribution to the variation in childhood intelligence.
Materials and Methods
Participants
We established the CHIC (Childhood Intelligence Consortium) to combine our efforts in elucidating the genetic and environmental bases of childhood intelligence. CHIC currently consists of six discovery (N = 12 441) and three replication (N = 5548) cohorts with a total sample size of 17 989 children of European ancestry for whom genome-wide SNP genotypes and intelligence scores are available (Table 1). The discovery cohorts are the Avon Longitudinal Study of Parents and Children (ALSPAC; N = 5517), the Lothian Birth Cohorts (LBC1921, N = 464; LBC1936, N = 947), the Brisbane Adolescent Twin Study, Queensland Institute of Medical Research (QIMR; N = 1752), the Western Australian Pregnancy Cohort Study (Raine, N = 936) and the Twins Early Development Study (TEDS; N = 2825). The three studies that formed the replication cohorts are the Generation Rotterdam study (Generation R, N = 1442), the Netherlands Twin Registry study (NTR; N = 739) and the University of Minnesota study (UMN; N = 3367). Each study obtained ethical approval from the relevant institution. The age of the children ranged between 6 and 18 years. More details about the cohorts are provided in the Supplementary Note of the Supplementary Online Information.
Table 1. Study characteristics of the discovery and replication samples.
| Cohort | Country | Platform | N | Intelligence measure | Mean age |
|---|---|---|---|---|---|
| Discovery samples (N = 12 441) | |||||
| ALSPAC-Bristol | UK | Illumina HumanHap550 | 5517 | WISC-III | 9 |
| LBC21-Lothians | UK | Illumina 610 Quad | 464 | Moray House Test No. 12 | 11 |
| LBC36-Lothians | UK | Illumina 610 Quad | 947 | Moray House Test No. 12 | 11 |
| QIMR-Brisbane | Australia | Illumina 610 | 1752 | MAB Full-scale IQ | 16 |
| Raine-Western Australia | Australia | Illumina 660 Quad | 936 | g score | 10 |
| TEDS-England/Wales | UK | Affymetrix 6.0 | 2825 | g score | 12 |
| Replication samples (N = 5548) | |||||
| Generation R-Rotterdam | The Netherlands | Illumina 610K | 1442 | SON-R 2,5–7 | 6 |
| NTR-Amsterdam | The Netherlands | Affymetrix | 739 | RAKIT, WISC-R, WISC-R-III, WAIS-III | 13 |
| UMN-Minnesota | USA | Illumina Human660W-Quad | 3367 | WISC-R, WAIS-R | 14 |
Abbreviations: IQ, Intelligence Quotient; QIMR, Queensland Institute of Medical Research.
Intelligence measure
We used the best available measure of general cognitive ability (g) or intelligence quotient (IQ), derived from diverse tests that assess both verbal and non-verbal ability (Table 1). In some studies, this was derived from an IQ-type test; in other studies, it was derived from the first unrotated factor of a factor analysis. Much research has shown that g is robust to the composition of the test battery.13 More details about the intelligence measure from each cohort are provided in the Supplementary Note.
Genotyping and quality controls
Individual sample quality control
Within each cohort, individuals were removed based on missingness, heterozygosity, relatedness, population and ethnic outliers, and other cohort-specific quality control (QC) steps. There were variations of QC between participating studies as the exact choice of QC thresholds depends on genotyping platform and study. More details on the QCs for each cohort are described in the Supplementary Note.
SNP QC
For the meta-analysis, SNPs were removed based on missingness (call rate <95%), minor allele frequency (<1%), Hardy–Weinberg (P-value <10−6), Mendelian errors (if family data were available) and other QC, such as the mean of GenCall score for Illumina arrays. As part of the QC procedure, we also calculated the average effective sample size (N) per cohort as a function of the allele frequency (p) and the standard error of the effect size (se) from the association test as where m is the number of SNPs and Rsq is the imputation quality score. This formula was derived from linear regression theory (Supplementary Note). This calculated N is a useful measure to check for the consistency of the reported sample size and the actual sample size that was used in the association analysis. We found that the calculated Ns were consistent with the reported Ns in all cohorts (Supplementary Table 1). We also checked for the consistency of the SNP allele frequencies between cohorts (Supplementary Figures 1 and 2).
Statistical analyses
Imputation
To facilitate the meta-analysis, the imputation of unobserved genotypes from the HAPMAP II CEU Panel (Release 22, NCBI Build36, dbSNP b126) was conducted within each cohort. This imputation was conducted on QC-ed data using the positive strand as the reference. We conducted the imputation using either BEAGLE,14 IMPUTE15 or MACH.16 We excluded imputed SNPs when the quality score (IMPUTE) or Rsq (MACH) was <0.3.
Association analysis
The association analysis was performed separately within each cohort. Except for the family data from QIMR, the analysis used the dosage score (the estimated counts of the reference allele in each individual; these estimates could be fractional and ranged from 0.0 to 2.0). An additive model was used on the standardised residuals (Z-score, transformed to normality if the phenotype is highly skewed) of the trait after adjusting for known covariates (age, sex, cohort, etc., including subtle population stratification effects, that is, the first four multi-dimensional scaling or PC (principal component) scores for each individual from a stratification analysis) on both genotyped and imputed SNPs. Both the directly genotyped and imputed SNPs were aligned to the HapMap reference strand. The Manhattan and Q–Q plots of the association P-values for each discovery cohort are presented in Supplementary Figures 3 and 4.
Meta-analysis
The results for associations between SNPs and childhood intelligence from the discovery samples were meta-analysed in the Metal package.17 We weighted the effect size estimates using the inverse of the corresponding squared standard errors. We also assessed the heterogeneity between the estimates in all cohorts using Cochran's Q statistic. The meta-analysis was performed for 2611179 SNPs. To avoid a disproportionate contribution of a single cohort to the results, we selected the association results for SNPs that survived QC in all cohorts (Total SNPs: 138093). The meta-analysis results from SNPs that survived QC in all cohorts were used for subsequent analyses, that is, gene-based analysis and profile scoring for the genetic prediction analysis. The detailed plot of the most significantly associated SNP in the meta-analysis is presented in Supplementary Figure 5.
Gene-based analysis
By considering all SNPs within a gene as a unit for the association analysis, a gene-based analysis can be a powerful complement to the single SNP–trait association analysis.18 We performed this gene-based analysis in Vegas software (Queensland Institute of Medical Research, Brisbane, Australia)18 using the P-values of the association between SNPs and childhood intelligence generated from the meta-analysis. We also conducted this gene-based analysis in each of the replication cohorts. Since there are ∼17000 genes, the genome-wide P-value threshold for declaring statistical significance following the Bonferroni correction for multiple testing was 0.05/17 000 = 3 × 10−6. Given the overlap between genes, the actual number of independent genes tested is likely to be smaller. Therefore, the Bonferroni correction for gene-based analysis is likely to be conservative.18 A detailed plot of the most significant gene from this gene-based analysis, FNBP1L, is presented in Supplementary Figure 6.
GCTA analysis
We estimated the contribution of all common SNPs on childhood intelligence by performing a linear mixed-model analysis to fit all genotyped SNPs simultaneously in the model, as implemented in the GCTA program.9 We excluded close relatives in the analysis by removing an individual from a pair where the estimated genetic coefficient of relatedness was > 0.025. One of the reasons for this exclusion is to eliminate bias due to common environmental factors. We conducted this analysis in the three largest cohorts, that is, ALSPAC, TEDS and UMN. The numbers of individuals used for this analysis were 5517, 2794 and 1736 children in the ALSPAC, TEDS and UMN cohorts, respectively.
Genetic prediction analysis
We used the estimates of SNP effect size from the meta-analysis to build a multi-SNP prediction model. We used this model to estimate the proportion of the phenotypic variation in independent samples that is due to genotypic information alone. To do this, we first identified independent SNPs from the meta-analysis using a P-value informed linkage disequilibrium (LD) clumping approach in PLINK19 with a cutoff of pairwise R2≤0.25 within a 200-KB window.20 Using this approach, we identified all independent SNPs that are significant at various P-value thresholds (Pt) (that is, 0.001, 0.005, 0.01, 0.05, 0.10, 0.25, 0.5 and 1). From groups of SNPs at each Pt threshold, we then calculated a quantitative genetic score19 or multi-SNP predictors in each of the three independent samples, that is, Generation R, NTR and UMN. We then performed a linear regression analysis between the quantitative genetic score and the observed measure of childhood intelligence, and quantified the precision of the predictor as the R2 measure of variance explained in the phenotype by the predictor.
Results and Discussion
To discover specific genetic variants affecting variation in childhood intelligence, we performed a meta-analysis of the SNPs–intelligence association results from the six discovery cohorts. Within each cohort, we tested for an association between (genotyped and imputed) SNPs that passed stringent quality controls and childhood intelligence, using an additive model. In our meta-analysis, we did not find any SNPs that reached a genome-wide significant P-value of 5 × 10−8, or, equivalent, a SNP that explained >0.24% of phenotypic variation (Figure 1). Q–Q plots and a genomic-control λ value of 1.078 are consistent with evidence for population stratification and/or polygenic variation.21 Population stratification appears unlikely because the phenotype was adjusted for at least the first four PCs or multi-dimensional scaling factors derived from population stratification analysis in each cohort (except for the Raine cohort). Therefore, the association results are consistent with many variants having small effects.
Figure 1.
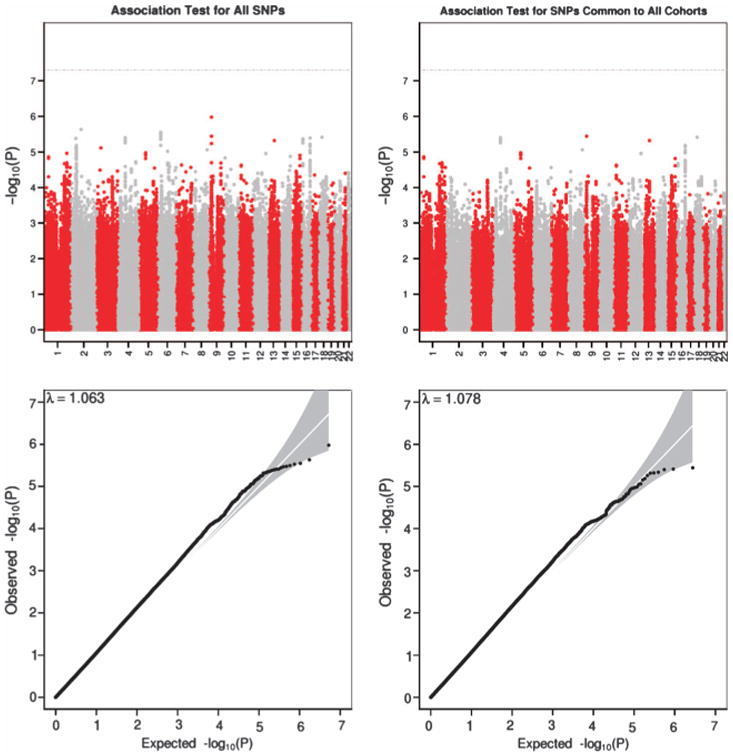
Manhattan and Q–Q plots of P-values of the association between single-nucleotide polymorphisms (SNPs) and Intelligence Quotient (IQ) in the discovery samples.
We conducted in silico replication for the top 100 independently significant SNPs identified from the meta-analysis of the discovery samples in three independent samples, the Generation Rotterdam Study (Generation R, N = 1442), the Netherlands Twin Register (NTR, N = 739) and the UMN Study (N = 3367). There was no SNP that reached nominal significance after a Bonferroni adjustment. When we meta-analysed the results from the discovery and replication cohorts, there were also no SNPs that reached a genome-wide significant threshold (Supplementary Table 2). However, when we plotted the estimated regression coefficients for the top 100 SNPs from the replication samples vs those from the discovery samples, we observed a positive correlation in Generation R (r = 0.340, P = 0.0005) and NTR (r = 0.219, P = 0.028), but not in UMN (r = −0.074, P = 0.46) samples (Supplementary Figure 7). These results demonstrated that the direction of the top 100 SNPs in the replication samples were generally consistent with those from the discovery samples. Again, these results are consistent with many variants with small effects.
To test whether the aggregate effect of a group of SNPs within a gene has a significant effect on childhood intelligence, we conducted a gene-based analysis18 and found that FNBP1L (formin binding protein 1-like) was the most strongly associated gene with childhood intelligence in the discovery samples (P = 4 × 10−5) (Table 2; Supplementary Table 3). This gene, previously known as Toca-1, encodes a protein that binds to both CDC42 and N-WASP and is involved in a pathway that links cell surface signals to the actin cytoskeleton.22 This is an interesting finding since a recent study also identified FNBP1L as the most significantly associated gene for adult intelligence from a gene-based analysis in ∼3500 individuals.6 A subset of our sample (that is, the Lothian Birth Cohorts, LBC 1921 and 1936) was part of the previous study6 that found the association between FNBP1L and adult intelligence. Therefore, we performed the gene-based analysis again by excluding LBC samples. FNBP1L was now ranked 80th with a P-value of 0.0031. The combined gene-based P-value for FNBP1L-childhood intelligence associations in all (discovery + replication), but excluding the LBC samples, was 0.014. These results are consistent with one or more causal variants in FNBP1L being associated with childhood intelligence in the population.
Table 2. Gene-based association analysis results for FNBP1L.
| Gene or SNP | Cohort | P-value |
|---|---|---|
| FNBP1L | Discovery | 0.00004 |
| FNBP1L | Discovery (excluding LBCs) | 0.0031 |
| FNBP1L | Discovery + replication (excluding LBCs) | 0.0137 |
| rs236330 (top SNP for FNBP1L) | Discovery | 0.00015 |
| rs236330 | Discovery (excluding LBCs) | 0.0018 |
| rs236330 | Discovery + replication (excluding LBCs) | 0.00045 |
Abbreviations: LBS, Lothian Birth Cohorts; SNP, single-nucleotide polymorphism.
Recently, we developed a method to estimate the contribution of common SNPs to the variation in complex traits using a linear model framework.9 We applied this method to estimate the contribution of all common SNPs to the variation of childhood intelligence by fitting all genotyped SNPs simultaneously in the model. This analysis requires substantial sample size to get estimates with small standard errors. Therefore, we performed this analysis separately in the three largest cohorts, ALSPAC, TEDS and UMN. The estimated proportions of the variation in childhood intelligence explained by common SNPs were 0.46 (s.e. 0.06), 0.22 (0.10) and 0.40 (0.21) for ALSPAC, TEDS and UMN cohorts, respectively (Table 3). These significant results imply that childhood intelligence is heritable and highly polygenic, and confirm and extend, to a different period in the human life course, previous reports on the polygenic nature of adult intelligence.3,6
Table 3. The proportion of the phenotypic variance of IQ explained by common SNPs (h2) estimated using the GCTA software9.
| Cohort | N | h2 (s.e.) | P-value |
|---|---|---|---|
| ALSPAC | 5517 | 0.46 (0.06) | 3.9 × 10−15 |
| TEDS | 2794 | 0.22 (0.10) | 0.014 |
| UMN | 1736 | 0.40 (0.21) | 0.028 |
Abbreviations: IQ, Intelligence Quotient; SNP, single-nucleotide polymorphism.
We investigated evidence for the polygenic nature of childhood intelligence further by building a multi-SNP predictor from the meta-analysis to predict childhood intelligence in three independent replication cohorts (Generation R, NTR and UMN). We found significant correlation between predictors and childhood intelligence in all replication cohorts (Figure 2; Supplementary Table 4). The maximum proportion of the phenotypic variance in childhood intelligence that was explained by genetic scores in each cohort were 1.2% (P = 6 × 10−5), 3.5% (P = 0.001) and 0.5% (P = 6 × 10−5) for Generation R, NTR and UMN, respectively. To verify whether these results were expected given the sample size, we performed the genetic prediction analysis on height and body mass index (BMI) using similar sample sizes. We used data from three population-based GWAS (the Health Professionals Follow-up Study (HPFS), the Nurses' Health Study (NHS) and the Atherosclerosis Risk in Communities (ARIC) study as the discovery set (N = 11 568 unrelated individuals)23 and a QIMR sample for validation (N = 3924 unrelated individuals).9 We found that the prediction accuracies for height and BMI were similar compared with that of childhood intelligence. Multi-SNP predictors explained 0.97% (P = 6.4 × 10−10) and 0.01% (P = 0.56) of variation in height and BMI in the independent sample, respectively.
Figure 2.
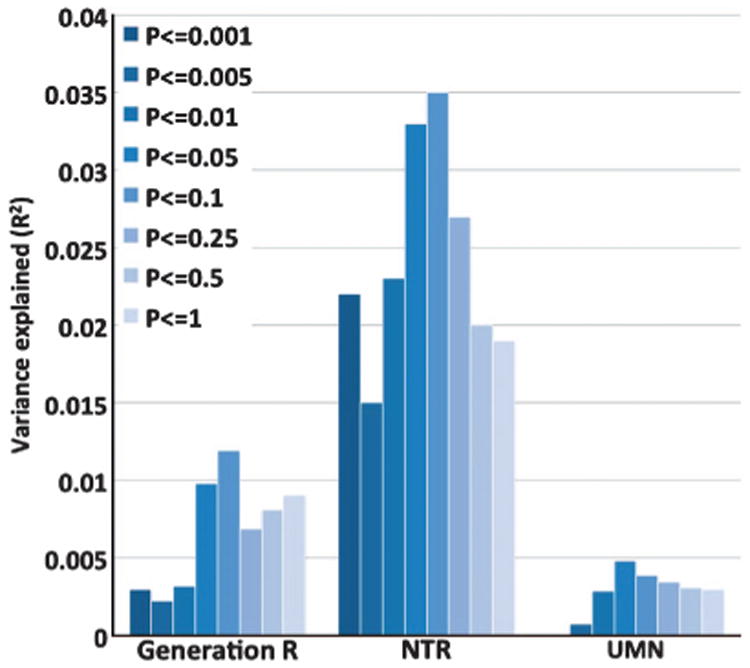
The proportion of the phenotypic variance in childhood intelligence explained by multi-single-nucleotide polymorphisms (SNPs) predictors (R2) in three replication samples. Each bar represents R2 for a given set of multi-SNPs predictors at a given P-value threshold.
In summary, while we did not find any individual SNP associated with childhood intelligence that reached genome-wide significance, the aggregate effect of common SNPs on the variation of childhood intelligence was significant in all three of our biggest samples. The proportion of phenotypic variance explained by common SNPs ranged from 0.22 to 0.46. These values are close to the heritability of 0.41 for childhood intelligence estimated from twin data.12 Since the estimates of total additive genetic variation from common SNPs are a lower limit of narrow-sense heritability,9,23,24 these results imply that there are many common causal variants with small effects segregating in the population, because rare variants are not in sufficient linkage disequilibrium with the genotyped and imputed SNPs to be captured by our whole-genome method. The multi-SNP prediction model was significant in all replication cohorts. Given the size of the discovery sample, the proportion of the childhood intelligence variation that can be explained by genetic predictors is consistent with previous findings on adult intelligence6 and with results from analyses on height and BMI. The variation in FNBP1L was significantly associated with childhood intelligence. This gene was previously associated with adult intelligence.6 Our results suggest that childhood intelligence is heritable and highly polygenic. Any attempt to identify individual genetic variants requires a larger sample size than the current study, consistent with other quantitative traits in human populations, including height,25 BMI,26,27 lipids,12 blood pressure28 and platelet count.29
Supplementary Material
Acknowledgments
We acknowledge funding from the Australian National Health and Medical Research Council (Grants 552498, 613672, 613601 and 1011506) and the Australian Research Council (Grant DP1093502). Funding support for the GWAS of Gene and Environment Initiatives in Type 2 Diabetes was provided through the NIH Genes, Environment and Health Initiative [GEI] (U01HG004399). The human subjects participating in the GWAS derive from The Nurses' Health Study and Health Professionals' Follow-up Study and these studies are supported by National Institutes of Health Grants CA87969, CA55075 and DK58845. Assistance with phenotype harmonisation and genotype cleaning, as well as with general study coordination, was provided by the Gene Environment Association Studies, GENEVA Coordinating Center (U01 HG004446). Assistance with data cleaning was provided by the National Center for Biotechnology Information. Funding support for genotyping, which was performed at the Broad Institute of MIT and Harvard, was provided by the NIH GEI (U01HG004424). The data sets used for the analyses described in this manuscript were obtained from dbGaP at [http://www.ncbi.nlm.nih.gov/sites/entrez?Db=gap] through dbGaP accession number [phs000091]. The Atherosclerosis Risk in Communities Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN26820 1100011C, and HHSN268201100012C), R01HL087641, R01HL59367 and R01HL 086694; National Human Genome Research Institute contract U01HG004402; and National Institutes of Health contract HHSN268200625226C. We thank the staff and participants of the ARIC study for their important contributions. Infrastructure was partly supported by Grant Number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research. BB and PMV are the recipients of the Australian National Health and Medical Research Council (NHMRC) fellowships. Acknowledgements for individual study cohorts are presented in the Supplementary Note.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
Author Contributions: IJD, GDS, RP and PMV designed the study and contributed to writing the paper. BB performed meta-analysis. BSP, OSPD, GD, NKH, M-JAB, RMK, RAMC, SF performed statistical analyses for each study cohort. BB and PMV wrote the first draft of the paper. Other authors contributed phenotypic and genotypic information on individual cohorts.
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
References
- 1.Deary IJ. Intelligence. Annu Rev Psychol. 2012;63:453–482. doi: 10.1146/annurev-psych-120710-100353. [DOI] [PubMed] [Google Scholar]
- 2.Deary IJ, Johnson W, Houlihan LM. Genetic foundations of human intelligence. Hum Genet. 2009;126:215–232. doi: 10.1007/s00439-009-0655-4. [DOI] [PubMed] [Google Scholar]
- 3.Chabris CF. Most reported genetic associations with general intelligence are probably false positives. Psychol Sci. 2011;23:1314–1323. doi: 10.1177/0956797611435528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Need AC, Attix DK, McEvoy JM, Cirulli ET, Linney KL, Hunt P, et al. A genome-wide study of common SNPs and CNVs in cognitive performance in the CANTAB. Hum Mol Genet. 2009;18:4650–4661. doi: 10.1093/hmg/ddp413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davies G, Tenesa A, Payton A, Yang J, Harris SE, Liewald D, et al. Genome-wide association studies establish that human intelligence is highly heritable and polygenic. Mol Psychiatry. 2011;16:996–1005. doi: 10.1038/mp.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Butcher LM, Davis OS, Craig IW, Plomin R. Genome-wide quantitative trait locus association scan of general cognitive ability using pooled DNA and 500K single nucleotide polymorphism microarrays. Genes Brain Behav. 2008;7:435–446. doi: 10.1111/j.1601-183X.2007.00368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis OS, Butcher LM, Docherty SJ, Meaburn EL, Curtis CJ, Simpson MA, et al. A three-stage genome-wide association study of general cognitive ability: hunting the small effects. Behav Genet. 2010;40:759–767. doi: 10.1007/s10519-010-9350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruano D, Abecasis GR, Glaser B, Lips ES, Cornelisse LN, de Jong AP, et al. Functional gene group analysis reveals a role of synaptic heterotrimeric G proteins in cognitive ability. Am J Hum Genet. 2010;86:113–125. doi: 10.1016/j.ajhg.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deary IJ, Whalley LJ, Lemmon H, Crawford JR, Starr JM. The stability of individual differences in mental ability from childhood to old age: follow-up of the 1932 Scottish Mental Survey. Intelligence. 2000;28:49–55. [Google Scholar]
- 12.Haworth CM, Wright MJ, Luciano M, Martin NG, de Geus EJ, van Beijsterveldt CE, et al. The heritability of general cognitive ability increases linearly from childhood to young adulthood. Mol Psychiatry. 2010;15:1112–1120. doi: 10.1038/mp.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson W, Bouchard TJ, Krueger RF, McGue M, Gottesman II. Just one g: consistent results from three test batteries. Intelligence. 2004;32:95–107. [Google Scholar]
- 14.Browning BL, Browning SR. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am J Hum Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genome-wide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu JZ, McRae AF, Nyholt DR, Medland SE, Wray NR, Brown KM, et al. A versatile gene-based test for genome-wide association studies. Am J Hum Genet. 2010;87:139–145. doi: 10.1016/j.ajhg.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, et al. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang J, Weedon MN, Purcell S, Lettre G, Estrada K, Willer CJ, et al. Genomic inflation factors under polygenic inheritance. Eur J Hum Genet. 2011;19:807–812. doi: 10.1038/ejhg.2011.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho HY, Rohatgi R, Lebensohn AM, Le M, Li J, Gygi SP, et al. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP-WIP complex. Cell. 2004;118:203–216. doi: 10.1016/j.cell.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, Manolio TA, Pasquale LR, Boerwinkle E, Caporaso N, Cunningham JM, et al. Genome partitioning of genetic variation for complex traits using common SNPs. Nat Genet. 2011;43:519–525. doi: 10.1038/ng.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Visscher PM, Yang J, Goddard ME. A commentary on ‘common SNPs explain a large proportion of the heritability for human height’ by Yang et al. 2010. Twin Res Hum Genet. 2010;13:517–524. doi: 10.1375/twin.13.6.517. [DOI] [PubMed] [Google Scholar]
- 25.Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010;467:832–838. doi: 10.1038/nature09410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gieger C, Radhakrishnan A, Cvejic A, Tang W, Porcu E, Pistis G, et al. New gene functions in megakaryopoiesis and platelet formation. Nature. 2011;480:201–208. doi: 10.1038/nature10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.