

Abstract
The nitrile group is an important functional group widely found in both pharmaceutical agents and natural products. More than 30 nitrile-containing pharmaceuticals have been approved by the FDA for the management of a broad range of clinical conditions in the last few decades. Incorporation of a nitrile group into lead compounds has gradually become a promising strategy in rational drug design as it can bring additional benefits including enhanced binding affinity to the target, improved pharmacokinetic profile of parent drugs, and reduced drug resistance. This paper reviews the existing drugs with a nitrile moiety that have been approved or in clinical trials, involving their targets, molecular mechanism of pharmacology and SAR studies, and classifies them into different categories based on their clinical usages.
This review summarizes versatile nitrile-containing drugs in the following aspects: target, mechanism of action and their structure activity relationship, and categorizes these drugs based on their target diseases.
1. Introduction
The nitrile group has played an increasingly important role in medicinal chemistry in recent decades. Since 2010, the United States Food and Drug Administration (US FDA) has approved at least one nitrile-containing drug every year, and reached the maximum number of five drugs in 2020 (Fig. 1). These marketed drugs with a nitrile moiety target a wide range of clinical disorders, including heart failure, hypertension, chronic myeloid leukemia, breast cancer, fungal infection, etc. Owing to the unique physicochemical properties of the nitrile substituent, nitrile-containing drugs may exert better pharmacokinetic and pharmacological effects compared with other drugs displaying similar therapeutic effects.
Fig. 1. The number of nitrile-containing drugs approved by the FDA from 2010 to 2020.

From the chemical perspective, the nitrile group contains a nitrogen atom with a free pair of electrons that links with an sp hybridized carbon atom, presenting a linear shape in space. The molecular volume of the nitrile group is only one-eighth the size of the methyl group, which together with the linear conformation are considered as the crucial reasons for fine accommodation in the binding pocket and great tolerance to various target mutations. Another remarkable benefit from the incorporation of the nitrile group into drug molecules is the improved pharmacokinetic profiles. Compounds containing the nitrile group generally have a lower clog P-value which indicates enhanced solubility. This changed physicochemical property also leads to obvious improvement in a series of pharmacokinetic parameters, involving increased system exposure, enhanced bioavailability, prolonged half-life time, etc.
Furthermore, the nitrile group is considerably metabolically stable and non-toxic. It usually remains unchanged when it is eliminated from the human body. Only in some extremely rare cases (i.e. mandelonitrile),1 toxic metabolites like cyanide may be released.
Studies2 have shown that there are mainly three types of interactions, hydrogen bonding, hydrophobic interactions and covalent contacts, that exist between the nitrile moiety of small molecule ligands and their target proteins. Among these interactions, hydrogen bonding is the major binding pattern for the nitrile group, which is firstly attributed to the strong electronegativity of the nitrogen atom of this moiety. Additionally, water molecules also can participate in the hydrogen bonding network, enabling the construction of a water-bridge for the association of nitrile with adjacent amino acid residues of the target, which in some cases are highly important for drugs to exhibit their binding activities. Furthermore, the nitrile substituent is commonly regarded as a bioisostere of carbonyl, hydroxyl, and halogen groups that universally act as hydrogen bond acceptors. Owing to its strong electron-withdrawing property, the nitrile moiety can greatly alter the electronic density of aromatic rings once it is attached directly to the benzene ring, potentially forming a π–π stacking interaction between the benzonitrile framework of a drug molecule and residues containing the aromatic system (such as phenylalanine, tyrosine, tryptophan and histidine) of a target protein. While the formation of covalent contacts is rare, a few examples like vildagliptin (25)3 are reported where the nitrile group can covalently bind to the active site of the target via the Michael addition reaction.
This paper has reviewed versatile nitrile-containing drugs recently approved or under clinical development in the following aspects: target, mechanism of action and their structure activity relationship, and categorizes these drugs based on their therapeutic areas. We also recommend readers to other excellent studies4–6 that had summarized previous work. We expect that this review could provide a useful reference to both medicinal chemists and organic chemists in the aspects of rational drug design and chemical synthesis.
2. Nitrile-containing drugs targeting cardiovascular system diseases
Potassium channels are proteins embedded on the cell membrane or mitochondrial inner membrane, regulating the inflow and outflow of potassium ions. The adenosine triphosphate potassium channel (KATP channel) is a well-known member of this huge potassium channel family, which is activated by a reduced intracellular ratio of ATP to ADP, and remains closed when this ratio rises. KATP channels are ubiquitously distributed on a variety of cells including cardiac muscle cells,7 neuron cells,8 and endocrine cells.9 Located on different kinds of cell types, there are distinct numbers and structures of KATP channels showing various biological functions. It is desirable to design more selective and potent drugs that target certain members of KATP channels. For example, cromakalim (1), targeting KATP channels on cardiac myocytes, is one of the lead compounds to perform the role of a KATP channel opener (KCO) for the treatment of patients with hypertension. More specifically, it is reported that this drug selectively opens the KATP channel, resulting in membrane hyperpolarization that inhibits Ca2+ and Na+ dependent ion channels on the same cell and eventually suppresses the ability of the cell to be excited. Unfortunately, cromakalim (1) was found to exert many adverse effects in clinic trials. Therefore, new compounds bearing the same chromane-6-carbonitrile scaffold with cromakalim (Fig. 2, 2–6) were pursued subsequently.10 Furthermore, incorporation of another type of chemical moiety, cyanoguanidine, in the drug scaffold, taking pinacidil (7) for example, was also proved to facilitate antihypertensive activity through activating the KATP channels. Of these KCOs, nitrile groups in their structure skeleton function as hydrogen bond acceptors to interact with the target protein.11 Additionally, they also result in electron-withdrawing effects to modify the electronic density of the aromatic ring (Fig. 2).
Fig. 2. The structures of cromakalim (1) and its derivatives.

Donitriptan (8), a potential drug under preclinical development, is a selective 5-hydroxytriptamine (5-HT)1D receptor agonist exerting inhibitory activity for capsaicin-induced vasodilation for the treatment of migraines (Fig. 3).12 In 2018, Garcia-Nafria's group disclosed the binding mode of donitriptan (8) with 5-HT1BR, which was determined by cryo-electron microscopy and single-particle analysis. It was reported that van der Waals forces as well as some polar contacts with Asp129 and Thr134 in the binding pocket were the dominant interactions existing between donitriptan (8) and 5-HT1BR, but the action of the cyano group remained unclear.13
Fig. 3. The structure of donitriptan (8).

Phenylalkylamines (PAAs) are a vital class of ligands targeting L-type calcium channels (LTCCs) that modulate the membrane permeability of calcium ions, which play a significant role in a diverse range of biochemical processes and are associated with various pathological conditions like arrhythmia.14 Chemically, PAA ligands are featured as two methoxylated benzene rings linked by a freely-rotating alkylamine chain with cyano and isopropyl moieties installed on the same chiral carbon (Fig. 4). Verapamil (9) was the earliest PAA to be developed as an LTCC inhibitor in the 1960s. Although it is marketed as a racemic drug for the management of arrhythmia, hypertension, and angina, its S enantiomer is far more potent than its R isoform. Besides verapamil (9), there are other two structure-related substances called devapamil (10) and gallopamil (11) that also can be used to block LTCCs via a similar molecular mechanism. In 2009, Tikhonov and coworkers15 firstly reported the importance of the cyano group in verapamil and its analogs on its inhibitory activities to LTCCs through molecular docking. It was shown that the nitrile group lied in the inner pore of this target and directly coordinated with the calcium ion bound to the glutamates on the selectivity filter. And this coordination stabilized the interaction between calcium and the selectivity filter, which could improve the inhibitory effects of PAAs.
Fig. 4. The structures of verapamil (9) and its derivatives.

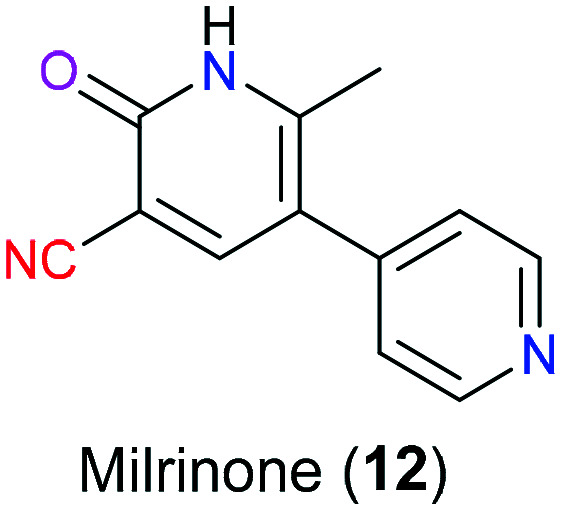
Milrinone (12) is a bipyridine derivative approved by the FDA in the management of congestive heart failure individuals who are refractory to pharmaceutical therapy or need inotropic support in cardiac transplantation (Fig. 5).16 Additionally, this drug also exhibits anti-inflammatory, cardioprotective, neuroprotective effects in clinical trials.17–19 As a selective low-Km cGMP-inhibited phosphodiesterase (also known as type III PDE or PDE3) inhibitor, milrinone (12) mimics cAMP to competitively occupy the binding site of PDE3 and hence increases the cAMP level in the cardiovascular system. In myocytes, the elevated cAMP promotes more calcium entry into cardiac myocytes and results in positive inotropic effects including myocardial contractility. Moreover, enhanced cAMP in vascular smooth muscle leads to peripheral vasodilation, which means reduced vascular resistance and increased cardiac output.20 It is also reported that besides acting as a PDE3 inhibitor, milrinone (12) was shown to antagonize the cardiac A1 receptor to display its inotropic properties.21 The X-ray crystal of the milrinone-PDE3 complex revealed its inhibitory action mode and the cyano moiety was hydrogen-bonded with residue His961.22
Fig. 5. The structure of milrinone (12).
Hereditary angioedema (HAE) is a rare, chronic disorder caused by autosomal dominant mutations in the serpin family G member (SERPING1) gene which encodes serine protease inhibitors such as complement 1 inhibitor (C1-INH), which is a significant regulatory molecule that balances the kallikrein–kinin system. This system starts with a stimulated activation of factor XII (FXII) to FXIIa, which can facilitate the proteolytic cleavage of prekallikrein to kallikrein that subsequently converts kininogen to bradykinin. The elevated bradykinin results in improved vascular permeability, increased extracellular fluid and therefore angioedema (Fig. 6). If C1-INH is insufficient, the enzymatic activity of both FXIIa and kallikrein will be abnormally increased, eventually leading to HAE disorder characterized by severe swelling of the skin, extremities, upper airway and gastrointestinal tract.23–25 In some cases, a potentially fatal condition would occur as a consequence of upper airway attacks, resulting in urgent demand for effective therapies for not only treatment of sudden attacks but also advanced prevention of this disease. In 2020, berotralstat (13), a highly selective kallikrein inhibitor developed by BioCryst Pharmaceuticals (NC, USA), was successfully approved by the FDA for prophylaxis of HAE attacks (Fig. 6). Unlike other treatment options involving ecallantide and icatibant that are administered through injection, this drug is a single daily dosing capsule with the advantage of acceptable tolerance and minimal adverse effects.26,27 In particular, berotralstat (13) is designed as therapy for type I HAE and type II HAE, which are related to CI-INH deficiency and dysfunction, respectively; however, the role of the nitrile group of berotralstat (13) molecule remains unclear.
Fig. 6. (a) The structure of berotralstat (13). (b) The molecular mechanism of action of berotralstat (13). FXII: factor XII; FXIIa: factor XIIa; C1 INH: complement 1 inhibitor.

3. Nitrile-containing drugs targeting endocrine system diseases
3.1. Drugs to treat endocrine organs
Prostate cancer always poses a huge threat to men's health due to its high incidence and mortality, and it is the second most universal male cancer in America, with more than 33 000 deaths in 2020.28 Consequently, much attention has been paid to finding out the specific molecular mechanism of this disease and developing effective therapies to treat it in the last few decades. It is believed that abnormal activation of the androgen receptor (AR) regulated by androgen is the major driving force for prostate tumor because AR as one of the transcription factors can stimulate cell growth-related gene expression.29 Apparently, AR is a pivotal target for rational drug design.
AR blocking agents include both steroidal and nonsteroidal substances. However, the former class of antiandrogen compounds has brought some unwanted adverse effects, which facilitates continuous update of nonsteroidal AR antagonists (Fig. 7) with better selectivity and pharmacokinetic profiles. Bicalutamide (14) is a first-generation drug widely used in combination with androgen deprivation therapy (ADT) for castration-sensitive prostate cancer (CSPC), which is an early stage of this disease. Unluckily, after about a two-year period of treatment, most patients would step into the castration-resistant prostate cancer stage (CRPC) caused by AR overexpression and/or relevant gene mutations, leading to a dramatic decrease of the pharmaceutical effects of first-generation AR antagonists. Accordingly, several next-generation drugs have been approved by the US FDA since 2012, including enzalutamide (15) (marketed in 2012), apalutamide (16) (marketed in 2018), and darolutamide (17) (marketed in 2019).30 Unlike bicalutamide (14), these drugs are full antagonists of AR and do not exhibit any agonist property. Furthermore, they have excellent pharmacokinetic parameters compared with the first-generation drugs, for example, better oral bioavailability, longer half-life time, and higher systematic exposure. In particular, darolutamide (17) with a unique structural scaffold is effectively blocked by the blood–brain barrier (BBB) resulting in fewer central nervous system-associated side effects. Also, it has a strong affinity for AR and can be tolerant to various mutants such as W74L, F876L, T877A, etc.31 Regardless of the numerous differences between the first-generation and the second-generation drugs, all of them have the similar moiety in their chemical structures, an electron-poor aromatic ring with a cyano substitution group, which is exactly the pharmacophore of antiandrogenic activity defined by the Tucker group in 1987.32 More specifically, nitrile is a strong electron-withdrawing group so it can decrease the electronic density of the benzene system to which it is attached, contributing to the ability to fit well in the AR binding environment.33 Taking bicalutamide (14) for example, the cyano group is embedded in the hydrogen bond network through directly interacting with the Gln residue of AR and indirectly being in contact with the binding-site water molecules. Additionally, the structure-related molecule enobosarm (18) is a nonsteroidal AR agonist instead of the aforesaid antagonist. And the major indication of this clinical candidate is to treat muscle wasting and impaired physical function in cancer patients.
Fig. 7. The structures of nonsteroidal androgen receptor antagonists.

Breast carcinoma continues to be a major health concern for females and ranks second behind lung cancer in terms of cancer-related mortality, causing approximately 25% of deaths of women suffering from this malignancy every year. It is reported that almost 70–80% of breast carcinoma has a very close relationship with the estrogen hormone level especially in postmenopausal women, which is also named as estrogen-receptor (ER) positive breast cancer.34 More specifically, estrogen is a pivotal factor to activate the predominant signalling pathway for cell growth and survival in the breast tumor through binding to the corresponding estrogen receptors. Hence, suppression of estrogen biosynthesis from androgen is one of the most effective and popular therapies for the treatment of this cancer. Anastrozole (19), letrozole (20) and fadrozole (21) are a newer generation of nonsteroidal aromatase inhibitors (NSAIs) which do not show undesired androgenic side effects such as weight gain, depression, and headache due to lack of steroidal structure.35 Therefore they exhibit better selectivity and less toxicity to combat ER-positive breast tumor compared with steroidal aromatase inhibitors (SAIs) (Fig. 8). As the molecular docking results suggested, anastrozole (19) and letrozole (20) possessed a similar binding fashion with the aromatase enzyme via reversible noncovalent interactions involving hydrogen bonding and π–π interaction. The cyano group mainly acted as an electron-withdrawing group to modify the electronic density of its attached benzene ring. Additionally, molecular docking simulation also suggested that their linear shape exactly fitted the steric space of the active site of aromatase, thus making them more selective than other drugs.36 Although anastrozole (19) and letrozole (20) have the same ligand–protein interaction pattern, there is still a difference in the binding affinity for specific CYP enzyme families. For instance, anastrozole (19) exhibits a potent inhibitory effect on CYP1A2, CYP2C8, and CYP3A4, but little or even no function on CYP2A6. In contrast, CYP2A6 can be effectively blocked by letrozole (20), while this drug plays an insufficient role in CYP3A4.37 In terms of fadrozole (21), it has not been approved by the US FDA (but launched in Japan now) and its molecular action with aromatase remains unclear.
Fig. 8. The structures of nonsteroidal aromatase inhibitors.

A wide range of crucial cellular processes involving proliferation, differentiation, angiogenesis, and migration are associated with signal transduction pathways that are regulated by the protein tyrosine kinase. Among those identified tyrosine kinases, HER (also known as ErbB) receptor tyrosine kinases have been indicated in various human cancers such as breast cancer, lung cancer, and other solid tumors, and have been extensively studied as anticancer drug targets. Generally, there are four members in the family of HER receptors, which are epidermal growth factor receptor (known as EGFR, HER1, or ErbB1), human epidermal growth factor receptor 2 (known as HER2, ErbB2 or neu), HER3 (ErbB3) and HER4 (ErbB4), respectively. These homologous proteins have similar constituents: a cysteine-rich extracellular ligand-binding domain, a single transmembrane lipophilic domain and a cytoplasmic tyrosine kinase domain. Their signal transduction pathways start with the binding of an extracellular ligand to them, which induces homodimerization or heterodimerization followed by autophosphorylation of the intrinsic tyrosine kinase domain. These phosphorylated residues provide docking sites for downstream signalling cascades, including the PI3K–AKT–mTOR pathway, Raf–MEK–ERK pathway and JAK–STAT pathway.38,39 However, none of the known ligands bind to HER2, and it is suggested that it invariably adopts an active conformation to be the heterodimerization partner of other HER receptors.40 Overexpression of HER2 accounts for 15–20% of all breast carcinomas and is correlated with poor prognosis.41 Neratinib (22), approved by the FDA in 2017, is an orally available and irreversible tyrosine kinase inhibitor against both early-stage and metastatic HER2-positive breast cancer, which occupies the ATP binding pocket of the tyrosine kinase region of HER2 via formation of a covalent bond between its Michael acceptor moiety and Cys805 in HER2.42 The cysteine residue is highly conserved among the HER family (Cys773 or 797 in HER1 and Cys805 in HER2); hence this drug is a pan-HER inhibitor with IC50 values of 92 nM for HER1, 59 nM for HER2 and 19 nM for HER4.43 Structure–activity relationship studies have shown that the basic dialkylamino group at the vicinity of the Michael acceptor not only served as an intramolecular catalyst for Michael addition but also increased its water solubility.44 The quinoline nitrogen of neratinib (22) interacted with the backbone NH of M769 and the carbocyano group was bioisosteric with the azomethine group of quinazoline combined with a water molecule, which was H-bonded to the sidechain OH of Thr830.45 Additionally, its derivative pyrotinib (23) was approved by the National Medical and Products Administration of China in 2018 for treatment of HER2 driven breast cancer, and other clinical trials in the management of gastric cancer have been investigated in both China and USA (Fig. 9).46
Fig. 9. The structures of neratinib (22) and pyrotinib (23).

3.2. Drugs to regulate hormone levels
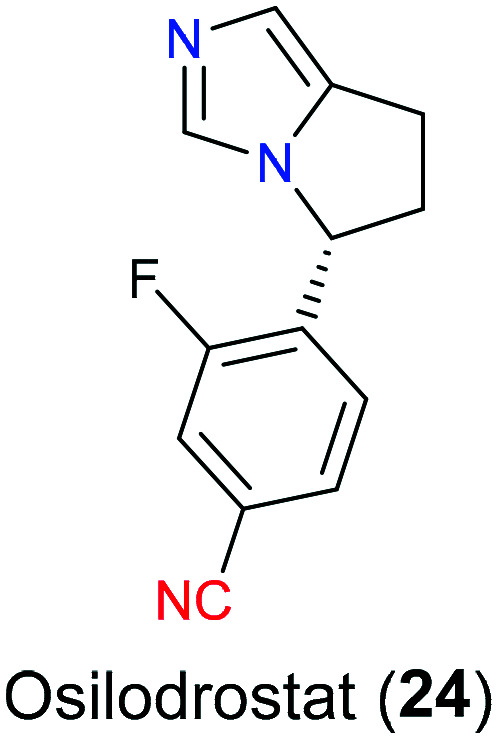
Cushing's disease (CD) is a serious endogenous metabolic disorder and more prevalent in women during their thirties to sixties, accompanied by symptoms of hypertension, psychological impairment and skeleton dysfunction. From the pathological perspective of CD, hypercortisolism caused by pituitary tumor disturbs the negative feedback via the hypothalamic–pituitary–adrenal axis.47 In a normal situation, the hypothalamus secrets corticotropin releasing hormone (CRH), which binds to the corresponding receptors on the pituitary gland to synthesize adrenocorticotropic hormone (ACTH) that subsequently acts on the adrenal glands to produce cortisol. Then cortisol circulates systematically to perform various bioactivities including blocking excess secretion of CRH and ACTH. However, for CD patients, ACTH continues to be released regardless of the negative feedback which finally leads to overproduction of cortisol.48 Osilodrostat (24), a novel potent medication, was recently approved in both EU and USA in 2020 as an adrenal steroidogenesis inhibitor that was applied for CD patients who are unable to tolerate transsphenoidal surgery (TSS) or have a reoccurring response to TSS (Fig. 10). As a inhibitor of enzyme 11-β-hydroxylase (CYP11β1), it blocked the last step of cortisol synthesis in the adrenal cortex.49 At first, osilodrostat (24) was developed by Novartis as a CYP11β2 (an aldosterone synthase) inhibitor. And its structural skeleton originated from fadrozole (21) which was an aromatase inhibitor used for treatment of breast cancer. Later, SAR studies50 have disclosed that this drug showed a potential inhibitory activity on CYP11β1, sharing 93% amino acid sequence identity with CYP11β2; hence it was repurposed for treatment of CD.
Fig. 10. The structure of osilodrostat (24).
Vildagliptin (25), saxagliptin (26) and alogliptin (27) are inhibitors of dipeptidyl peptidase IV (DDP-4is) used for the treatment of type 2 diabetes mellitus (T2DM), which is a prevalent world-wide disease characterized by hyperglycemic condition, with an estimated 422 million cases in 2020 (Fig. 11). Currently, DPP-4is are regarded as the second/third-line therapy for T2DM with less toxicity than other antidiabetic medicines, and they always play an extremely significant role in the occasion when the first-line drug, metformin, is ineffective for patients.51 As the name indicates, the major task of this category of glucose-lowering drugs is to block DPP IV enzymes which have the ability for enzymatic hydrolysis of incretins secreted by the gut including glucagon-like peptide-1 (GLP-1) and gastric insulinotropic polypeptide (GIP). These gut hormones can speed up the release of insulin from pancreatic β cells after food intake.52 Hence, DPP-4is can effectively elevate the insulin secretion and control the blood glucose level via inhibiting DPP IV activity and therefore prolonging the half-life of these hormones. From the view of the chemical structure, vildagliptin (25) and saxagliptin (26) share a very similar skeleton showing a 2-cyano pyrrolidine. SAR analysis indicated that this nitrile group not only contributed to their inhibitory properties but also maintained excellent stability for the oral drug delivery route. In 2007, Peters53 demonstrated that the nitrile group reversibly formed a covalent bond with Ser630 to generate an imidate adduct in the active site of DDP, which was particularly important for their inhibitory action. Unfortunately, the nitrile moiety in vildagliptin (25) still faces the risk of a spontaneous intramolecular cyclization event to afford an inactive compound in solution. However, saxagliptin (26) developed by Bristol-Myers Squibb overcomes this issue through the construction of a cyclopropyl ring fused directly with the 2-cyano pyrrolidine and therefore exhibits more robust therapeutic effects and better pharmacokinetic profiles than vildagliptin. Another selective oral DPP-4i is alogliptin (27), approved in 2013 by the FDA, which has a different chemical model from vildagliptin (25) and saxagliptin (26), but has a similar purpose for the introduction of the cyano moiety in the initial drug design stage.54
Fig. 11. The structures of dipeptidyl peptidase IV inhibitors.

4. Nitrile-containing drugs targeting lymphatic system diseases
4.1. Drugs to treat lymphoid cells
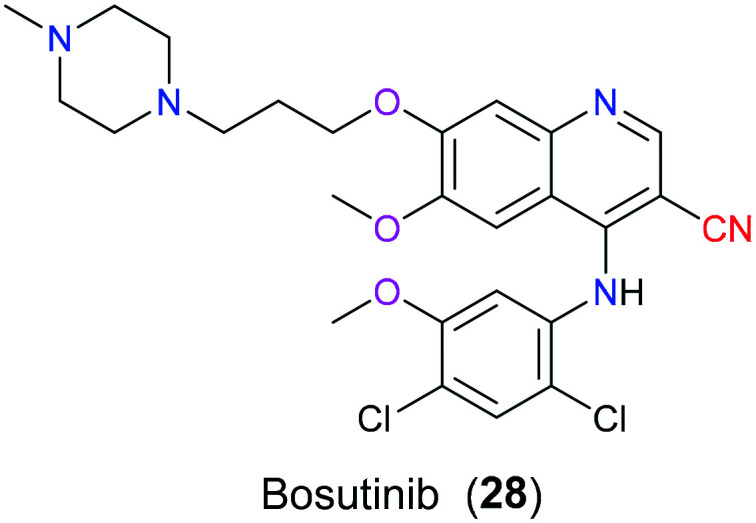
Chronic myeloid leukemia (CML) is the most common disease appearing among older adults over 65 years. The natural history of this myeloproliferative neoplasm characterized by the occurrence of Philadelphia chromosome (Ph+ CML) encompasses the following stages: chronic phase (CP), accelerated phase (AP), and blast phase (BP). The abnormal Ph chromosome contains a fused gene called BCR-Abl that is capable of being translated into a gain-of-function product, an overactive tyrosine kinase complex. Therefore, BCR-Abl tyrosine kinase is a valuable target for the treatment of Ph+ CML.55 In 2001, imatinib (Gleevec) was successfully approved by the FDA as a first-generation tyrosine kinase inhibitor (TKI) for the treatment of this disease. Although this drug is effective enough to convert this horrible cancer with a median survival period of 3–5 years to a chronic disease, increasingly developed imatinib-resistance attracts attention to exploring newer generations of TKIs.56 Consequently, bosutinib (28), a more potent dual Abl/Src inhibitor, was approved by the FDA in 2012 which is mainly used for the treatment of patients with multiple imatinib resistance Ph+ CML (Fig. 12). Molecular docking mode has reported that the nitrile group in bosutinib (28) is in close contact with T315 and V299 of Abl protein via van der Waal force, which offered a reasonable explanation for the ineffectiveness to T315I and V299L mutants. Additionally, studies showed that nitrile-containing TKIs could be used to further develop more selective kinase inhibitors based on their difference in electrostatic properties which play a vital role in differentiating ATP-binding sites with highly conserved sequence.57
Fig. 12. The structure of bosutinib (28).
Recently, medicinal chemists have paid growing attention to the hedgehog (Hh) signalling pathway due to its close relation to the pathogenesis of various cancers, especially acute myeloid leukemia (AML), basal cell cancer, and medulloblastoma carcinoma. From the perspective of the detailed molecular mechanism of this signalling pathway, Sonic hedgehog (SHh), Indian hedgehog (IHh) and Desert hedgehog (DHh) are three major ligands binding to their Patched receptor (Ptch). Following this, the receptor transduces the signals from the ligand to the downstream second messengers, which ultimately results in the activation of Gli1, Gli2, and Gli3, which are the three glioma-dependent transcription modulators that can regulate the proliferation-related gene expression. Hence, if this Hh signalling pathway is activated mistakenly, overexpression of certain genes will lead to the occurrence of some cancers, CML, for example.58 Smoothened (Smo), a seven-transmembrane protein, acting as a key mediator actively participates in the above downstream signalling pathway and has attractive potential to be a drug target for regulation of the aberrant Hh signalling transduction pathway. In 2018, the FDA approved glasdegib (29), a small molecule inhibitor of Smo protein, to suppress the overactive Hh signal pathway in patients with AML (Fig. 13). SAR studies59 disclosed that the cyano group installed on the para position of the aromatic ring could be beneficial to a satisfying pharmacokinetic profile, referring to the enhanced hydrophilicity and metabolic stability.
Fig. 13. The structure of glasdegib (29).

Isocitrate dehydrogenase (IDH) is a key enzyme that plays an active role in the Krebs cycle via the catalytic oxidative decarboxylation of isocitrate to carbon dioxide and affords α-ketoglutarate (α-KG) which engages in versatile cellular processes including DNA demethylation and adaption to hypoxia through binding to α-KG-dependent dioxygenases.60 Somatic point mutations at a pivotal arginine residue (R132) within the active site of IDH1 and R140 or R172 within the active site of IDH2 confer a neomorphic gain-of-function activity, leading to the reduction of α-KG to generate the oncometabolite d-2-hydroxyglutarate (2-HG) which shares a very similar chemical structure with α-KG and thereby competitively inhibits α-KG-dependent dioxygenases, thus inducing impaired cellular differentiation and epigenetic deregulation. These hotspot mutations have been found in multiple hematologic malignancies and solid tumors, including cholangiocarcinoma and acute myeloid leukemia (AML). It is reported that approximately 6–10% of patients with AML show an elevated mutant IDH1(mIDH1) dependent 2-HG level.61 Ivosidenib (30) is a first-in-class, orally available, potent, targeted, allosteric inhibitor of mIDH1 enzyme, which received US FDA approval in 2018 for treatment of patients with newly diagnosed AML who are at least 75 years old or who have comorbidities precluding the use of intensive induction chemotherapy, and patients with relapsed or refractory (R/R) AML (Fig. 14).62 The X-ray model showed that ivosidenib (30) exerted its inhibitory activity through binding to the allosteric sites and hence stabilizing the open and inactive conformation of mIDH1.63 SAR studies had reported that the discovery of ivosidenib (30) could trace back to lead optimization of AGI-5198 (31), and replacing the imidazole group with a pyrimidine ring on the oxidized proline moiety greatly improved the metabolic stability. Additionally, the substitution of the nitrile group with the fluorine group slightly impaired the inhibitory potency.64 Other therapeutic utilities of ivosidenib (30) including treatment of patients with advanced/metastatic cholangiocarcinoma have been investigated recently.65
Fig. 14. The structures of ivosidenib (30) and its derivative (31).

4.2. Drugs to treat lymphatic organs
Janus kinases (JAKs) belong to the big family of tyrosine kinases that play a vital role in modulating intracellular signal transduction involving the expression level of cytokine and growth factor, hematopoiesis and immune function.66 More specifically, endogenous factors such as cytokine and chemokine bind to the corresponding receptors and therefore activate JAKs which are associated with the intracellular domain of these receptors. Following, JAKs switch on the signal transducers and activators of transcriptions (STATs) via phosphorylation of certain amino acid residues, which is known as the JAK/STAT signalling pathway that eventually induces a series of various biological sequences.67 Ruxolitinib (32), tofacitinib (33), and baricitinib (34) are JAK inhibitors that possess a nitrile group in their chemical structures (Fig. 15). In particular, the first one approved by the FDA among them is ruxolitinib (32), which was marketed for the treatment of myelofibrosis in 2011, and then polycythemia vera and acute graft-versus-host disease as two other indications were approved in 2014 and 2019, respectively.68 As a potent and orally administered JAK1/JAK2 inhibitor, it is surprising to find that ruxolitinib (32) was likely to be considered in the treatment of COVID-19, a severely global pandemic viral infection, through blocking the JAK/STAT signalling pathway.69 In terms of tofacitinib (33), the FDA approved it for methotrexate-resistant patients with rheumatoid arthritis (RA) in 2012, and this medicine exerted its pharmacological effects via suppressing JAK1/JAK3 function. Baricitinib (34) is a newly developed JAK1/JAK2 inhibitor (approved in 2018) by Eli Lilly to be mainly used for inflammatory conditions. SAR analysis70 disclosed that ruxolitinib (32) reversely and selectively bound to the corresponding JAK enzyme via noncovalent interaction involving hydrogen bonding and hydrophobic interaction. In particular, its carbonitrile moiety participated in van der Waals hydrophobic interactions with D994 and N981.
Fig. 15. The structures of Janus kinase inhibitors.

4.3. Drugs to treat autoimmune condition
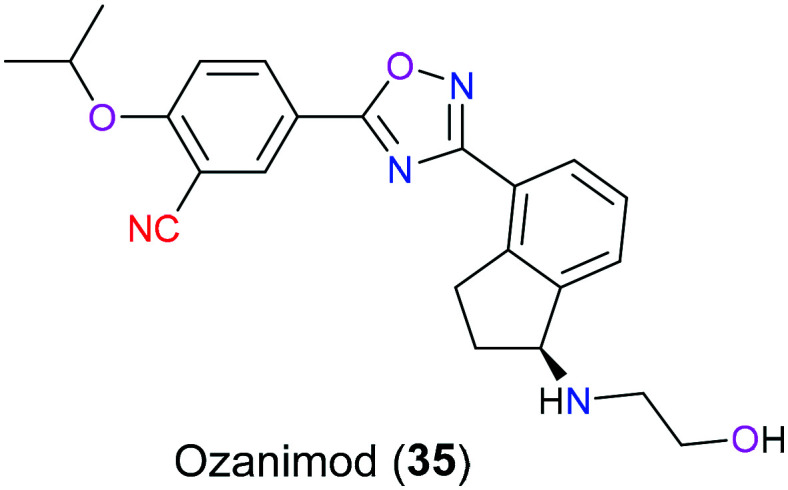
Multiple sclerosis (MS) is a chronic autoimmune disease of the central nervous system characterized by demyelination, inflammation, disruption of the blood–brain barrier, and neurodegenerative symptoms such as neuroaxonal loss and progressive atrophy. There are four types of MS: clinically isolated syndrome, relapsing–remitting MS (RRMS), secondary progressive MS and primary progressive MS. Of these phenotypes, RRMS is the most prevalent which accounts for approximately 85% of individuals with MS, which is characterized by a period of remission after episodes of relapses.71 Sphingosine 1-phosphate (S1P) as a kind of lipid molecule plays a pivotal role in many physiological and pathophysiological events including MS, inflammation, and cancer via versatile G protein-coupled receptor (GPCR). Therefore, there is a wide range of investigations associated with S1P modulators that engage in S1P signalling cascades to treat the corresponding disorders. Ozanimod (35), a selective S1P receptor modulator, was approved in 2020 for the treatment of RRMS with a favorable benefit–risk profile and superior selectivity compared to the first generation S1P receptor modulator, fingolimod (Fig. 16). This is mainly because only S1PR1 and S1PR5 are bound to ozanimod (35), but for fingolimod, more than 4 subtypes of S1PR are its targets.72 The underlying biochemical mechanism for ozanimod (35) begins with activation of S1P receptor on lymphocytes via binding to it, resulting in reduction of the number of lymphocytes through preventing them from migrating from lymphoid tissue and decrease of cytokine receptors on lymphocytes.73 Although there are some descriptions about the pharmacology and metabolism of ozanimod (35), the specific molecular mechanisms still remain complex and ambiguous.74 In 2021, Thomas et al.75 studied the SAR of ozanimod (35) by a molecular docking approach. Docking results indicated that there were five hydrogen bond interactions, one ionic interaction, and three hydrophobic interactions between ozanimod (35) and the protein target, and the nitrile group was hydrogen-bonded with binding-site residue Thr117.
Fig. 16. The structure of ozanimod (35).
4.4. Anti-inflammatory drugs
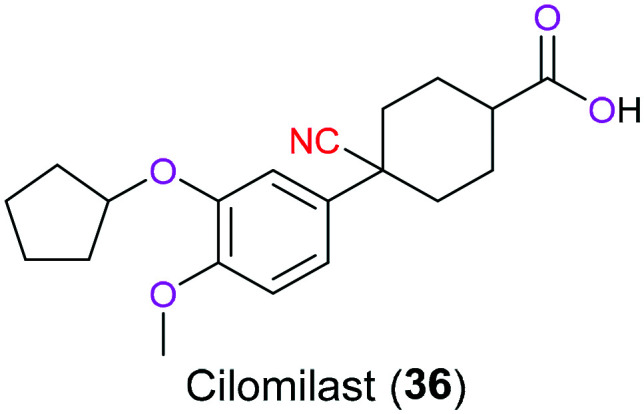
Inhibitors of phosphodiesterases (PDEs) possess promising therapeutic potential for the treatment of various inflammatory conditions including asthma and chronic obstructive pulmonary disease (COPD) via suppression of overexpressed innate and acquired immunity. This is because the blockage of PDEs can inhibit enzymatic hydrolysis of several second messengers and therefore elevate their intracellular concentration, especially for cyclic 3′,5′-adenosine monophosphate (cAMP) and 3′,5′-guanosine monophosphate (cGMP), which play a critical action in a myriad of physiological processes involving immune responses through certain signalling pathways associated with a cascade of protein kinase activation and/or gene expression.76 Among numerous PDE inhibitors, cilomilast (36) developed by GlaxoSmithKline as a next-generation PDE4 inhibitor in clinical phase III is designed to treat COPD that is characterized by many inflammatory conditions such as frequent breathlessness and increased mucus excretion (Fig. 17). Although having both bronchodilation and anti-inflammatory properties, a narrow therapeutic window and toxicity to the central nervous system limit the application of this drug.77 In 2004, Zhang et al.78 demonstrated the structural basis for its inhibitory activity on PDE4. In terms of the role of the nitrile part in its structure, it mainly participated in the formation of hydrogen bonds with M347, L393, and F414 in the metal binding pocket (M), explaining why cilomilast (36) was more effective than other PDE4 inhibitors (filaminast and mesopram). In 2020, El-Azab' group79 reported that suppression effects on PDE4B could be enhanced by the introduction of a cyanopyrimidine fragment in the 2-cyclopentyloxyanisole skeleton.
Fig. 17. The structure of cilomilast (36).
Leflunomide (37) is an orally available, disease-modifying isoxazole derivative with good anti-inflammatory, anti-cancer, and immunosuppressive activities approved as an antirheumatic drug by US in 1998 and by EU in 1999, respectively.80 Recently, some investigations have been conducted to explore its pharmacological potential in the treatment of multiple myeloma and neuroblastoma.81,82 After administration, leflunomide (37) will be rapidly and almost completely transformed to its active ring-opening metabolite teriflunomide (38) in the gastrointestinal tract and liver. Teriflunomide (38) exists in Z and E isoforms which can be interconverted through a keto form (Fig. 18). Due to the presence of an intramolecular hydrogen bond, the Z isomer is energetically favored than the E isomer, whereas this hydrogen bond interferes with the binding affinity of teriflunomide (38) with its target protein.83 In 2012, the FDA approved teriflunomide (38) for management of relapsing forms of multiple sclerosis. Teriflunomide (38) exhibits its biological function through inhibition of human dihydroorotate dehydrogenase (DHODH), a mitochondrial enzyme participating in the rate-limiting step in the pyrimidine de novo biosynthesis pathway. Once it reversibly inhibits DHODH, lymphocyte proliferation will be halted owing to the shortage of the necessary building block pyrimidine for nucleic acid synthesis, thus resulting in immunomodulator effects.84,85 The SAR and X-ray crystal studies have shown that amide carbonyl indirectly forms a hydrogen bond with amino acid Arg136 of DHODH via a water molecule, and the enolic hydroxyl group is hydrogen-bonded directly with Tyr356 in the active pocket. Additionally, the 4-trifluoromethylphenyl group is in contact with this enzyme through essential hydrophobic interactions in the tunnel. The cyano moiety does not exhibit a fixed binding pattern but plays a crucial role in receiving external interactions.86,87
Fig. 18. The metabolic conversion from leflunomide (37) to (Z)-teriflunomide (38) and (E)-teriflunomide (38).

Gout is an increasingly common inflammatory disease characterized by crystallization and deposition of monosodium urate crystals in or around a joint.88 Hyperuricemia, defined as the concentration of serum uric acid (sUA) greater than 6.8 mg dL−1, is thought to be the underlying cause of gout, resulting from overproduction or underexcretion of uric acid. In addition to gout, hyperuricemia also leads to renal disease and cardiovascular disorders such as hypertension and hyperlipidemia.89 Currently, interruption of uric acid biosynthesis is one of the most prevalent treatments of gout through inhibiting xanthine oxidoreductase (XOR) which actively participates in the cascade of hypoxanthine to xanthine and then to the end product uric acid.90 XOR includes xanthine oxidase (XO) and xanthine dehydrogenase (XDH), which share great similarity in biochemical structure and catalytic function, and can interconvert mutually. Briefly speaking, the major biological activity of XOR enzyme is to transfer oxygen from water to a wide range of cellular substrates such as purine, pterin and aldehyde. During this process, electrons initially transfer from substrates to the Moco redox center (from Mo(vi) to Mo(iv)) of the enzyme, and then to flavin adenine dinucleotide (FAD) cofactor via Fe2S2 clusters. For XO enzyme, electrons eventually migrate from FAD to O2 resulting in H2O2/O2−, and for XDH enzyme, NAD+ is the last accommodation for these two electrons and corresponding NADH is produced.91 Febuxostat (39) is a second generation, non-purine, selective, and structure-based XOR inhibitor approved for treatment of gout. And topiroxostat (40) is a third-generation XOR inhibitor used as an alternative for patients who develop resistance to febuxostat (39), and this drug is still under investigation (Fig. 19).92 In 2003, Nishino and co-workers93 disclosed the inhibitory mechanism of febuxostat (39) via the X-ray crystal structure of the XDH–febuxostat complex. This drug was bound in the narrow channel leading from the bulk solvent to the inner Mo-pterin cofactor, and acted like a plug to inhibit the entrance of the purine substrate. The CN group in this drug was hydrogen-bonded to Asn768 and this interaction not only stabilized the position of the phenyl group, but also contributed to the binding. Studies showed that if the CN group was removed, the binding affinity would dramatically decrease. As for topiroxostat (40), another research reported94 that its trihydroxy metabolite was found to chelate with the Mo(vi) center, although the specific orientation in the binding site was still unclear.
Fig. 19. The left panel shows the structures of febuxostat (39) and topiroxostat (40); the right panel shows the hypoxanthine metabolism catalyzed by XOR. XO: xanthine oxidase; XDH: xanthine dehydrogenase; NAD+: nicotinamide adenine dinucleotide; NADPH: dihydronicotinamide adenine dinucleotide.

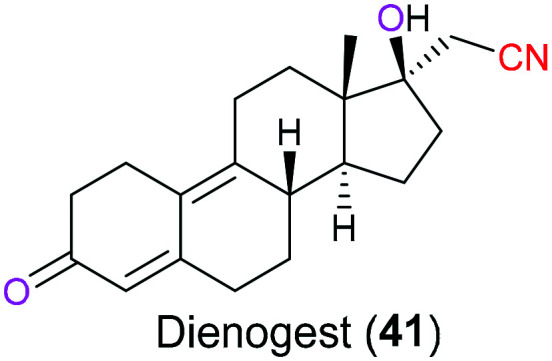
Dienogest (41) is an orally administered progestin derived from C19-nortestosterone, which has been combined with other drugs for the treatment of hormonal contraception and hormone replacement therapy for many years (Fig. 20). In 2011, the US FDA initially licensed this drug independently for treatment of symptomatic endometriosis which is an inflammatory disease characterized by endometrial lesion.95 Dienogest (41) has a unique chemical structure that endows it with therapeutical effects of both C19-nortestosterone and progesterone derivatives, which result in enhanced bioavailability and reduced side effects on metabolic and cardiovascular systems, respectively. It is the olefin in the steroidal B ring that is responsible for its progesterone activities. Additionally, incorporation of a cyanomethyl moiety at the C-17 position will cause less liver toxicity compared with other C19-nortestosterone derivatives that usually have a typical ethinyl group at C-17.96
Fig. 20. The structure of dienogest (41).
Atopic dermatitis (AD), also known as eczema and atopic eczema, is a common chronic inflammatory skin disorder affecting 15% to 20% of young children and approximately 3% of adults worldwide, which is clinically characterized by intense pruritus, eczematous rashes, and dermatitis plaques and usually associated with allergy and asthma.97 Crisaborole (42) is a nonsteroidal anti-inflammatory topical ointment firstly approved by the FDA in 2016 for treatment of mild-to-moderate AD in patients aged ≥2 years. Furthermore, a supplemental new drug application to loosen the age limit to accommodate patients ≥3 months of age was also received by the FDA in March 2020.98 The pharmacology of crisaborole (42) is to act as a small molecule selective inhibitor for phosphodiesterase 4 (PDE4) enzyme which balances the cellular cAMP level via its hydrolytic function. Consequently, PDE4 inhibition can increase the cAMP level, which in turn suppresses the production of pro-inflammatory cytokines such as interleukin (IL)-2, IL-4, IL-5, IL-10, IL-13, IL-17, interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α), eventually resulting in the relief of AD symptoms.99 SAR studies have implicated that the boron atom in crisaborole (42) and its derivative (AN2898) (43) adopted a tetrahedral configuration to act as a bioisostere of phosphate, and hence exhibited their inhibitory activity through coordination with the water molecule activated by the bimetal ions (zinc and magnesium) in the active site of PDE4.100 The cyanophenoxyl moiety was located at the entrance of the hydrophobic pocket mainly formed by residues Phe446, Ile410, and Gln443, and CN as an electron-withdrawing group and hydrogen bond acceptor contributed to the enhanced inhibitory activity.101 Further modification of crisaborole (42) focused on installation of bicyclic scaffolds encompassing electron-withdrawing groups to replace cyanophenyl to deeply probe the hydrophobic pocket, exemplified by compound 44 (Fig. 21).102
Fig. 21. The structures of crisaborole (42), AN2898 (43), and 44.

4.5. Antibacterial drugs
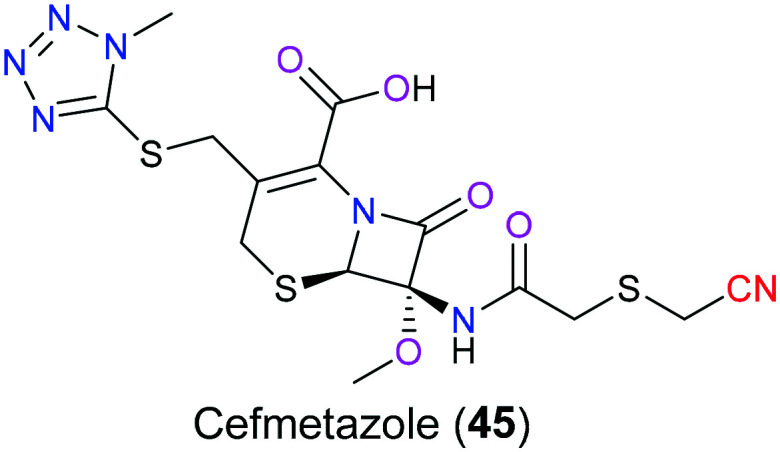
Cefmetazole (45) is an antibiotic targeting caseinolytic protease subunit P (ClpP), which is a highly conserved serine protease in microbes and plays an extremely significant role in the hydrolysis of intracellular proteins in bacteria via its proteolytic chamber (Fig. 22). Based on its proteolytic function, ClpP takes a very active part in necessary biological activities that maintain bacterial growth and survival, such as cleaning up wrongly folded peptides during cell division. Studies have shown that Legionella pneumophila with suppressed ClpP has lower infectiousness. Therefore, cefmetazole (45), one of the ClpP inhibitors, is expected to display an excellent antibacterial activity, especially on Gram-negative bacteria. SAR studies indicated103 that there was an additional covalent bond formed via the nitrile group with T186 in the binding pocket, which enhanced its therapeutic effects compared with other structure-related drugs without a nitrile moiety.
Fig. 22. The structure of cefmetazole (45).
Finafloxacin (46) is one of the fourth-generation fluoroquinolone antibiotics characterized by a unique chiral 7-pyrrolo-oxazinyl substituent and an 8-cyano group (Fig. 23), which was initially approved by the US FDA in 2014 as an otic suspension to treat swimmer's ear caused by Pseudomonas aeruginosa and Staphylococcus aureus. Nowadays, new indications including treatment for urinary tract infections and Helicobacter pylori infections are being developed by MerLion Pharmaceuticals.104 The antimicrobial activity of finafloxacin (46) includes a straightforward blockage of microbial DNA synthesis through the formation of a ternary complex of this drug with DNA strands and bacterial topoisomerase type II enzymes (DNA gyrase and topoisomerase IV), which interrupts bacterial DNA replication and transcription and finally cause bacterial death. Unlike other generation fluoroquinolones which display a preferred affinity for one of the topoisomerase type II enzymes, finafloxacin (46) inhibits them with similar potency. This dual inhibitory activity makes this drug less susceptible to enzyme mutation and therefore drug resistance is hard to be developed.105 Furthermore, it is reported that finafloxacin (46) has resistance against efflux pumps, the bacterial multidrug transporter NorA, which indicates an enhanced bioavailability compared with other fluoroquinolones.106 In addition, another remarkable advantage is that this compound exhibits the maximum bacterial inhibitory activity under acidic conditions (pH 5–6) where other fluoroquinolones are inactivated. Medicinal chemists could make the best use of this unique property to combat infections occurring at acidic body sites including the skin, respiratory system and urinary tract.107 The nitrile moiety in this drug acts as a pseudohalogen to enhance its oral pharmacokinetics, to enlarge the antimicrobial spectrum by the inclusion of anti-Gram positive bacterial and anti-anaerobe activities, and to reduce drug resistance.108–110 Pradofloxacin (47) is another fluoroquinolone with a 8-cyano substituent developed against both aerobic and anaerobic bacteria infecting dogs and cats, which shares an extremely similar chemical pattern to finafloxacin (46) (Fig. 23).111
Fig. 23. The structures of finafloxacin (46) and pradofloxacin (47).

4.6. Antifungal drugs
As the populations of immunocompromised patients due to the lack of medicinal therapy for AIDS, hematopoietic malignancies, and organ transplantations increase, invasive opportunistic fungal infections have been an increasing healthcare problem associated with high mortality and morbidity.112 In 2015, isavuconazonium sulfate (49) as a second-generation water-soluble triazole was launched in the U.S. and Europe for oral and intravenous treatment of invasive aspergillosis and mucormycosis, which are two of the most common pathogens causing invasive fungal infections. Besides that, studies113,114 also demonstrated that isavuconazonium sulfate (49) was an extended-spectrum antifungal agent in vitro against versatile yeasts and molds such as Fusarium spp., Candida spp., and Cryptococcus spp. and black yeast. From the pharmacokinetic perspective of this prodrug, it will be metabolized rapidly into isavuconazole (48) (its active moiety) through cleavage of an ester bond that links triazolium salt with the aminocarboxyl (N-(3-acetoxypropyl)-N-methylamino-carboxymethyl) substituent.115 Structurally similar to isavuconazole (48), ravuconazole (50) is an investigational antifungal agent exhibiting excellent inhibitory efficacy against a broad range of fungal species such as Aspergillus and Candida. To enhance the bioavailability and solubility of ravuconazole (50), fosravuconazole l-lysine ethanolate (E1224) (51) was clinically developed in 2007 (Fig. 24). Both isavuconazole (48) and ravuconazole (50) showed a superior safety and pharmacokinetic profile, which specifically refers to high affinity with the target, low clearance and high serum concentration.116,117 Similar to other triazoles, their mechanism of action is to interrupt ergosterol biosynthesis which is a vital fungal cell membrane component through inhibition of cytochrome P450-dependent lanosterol 14α-demethylase encoded by gene Erg11p in yeast and CYP51 in filamentous fungi. This disruption results in aberrant sterol accumulation and thereby disorders in the fungal membrane structure array and function. In detail, the triazole ring of these two drugs interacts with the heme moiety of lanosterol 14α-demethylase in the active site, and the side arm determines the inhibitory activity against different pathogens and relates to drug resistance.118 The molecular docking analysis of the ravuconazole–lanosterol 14α-demethylase complex showed that 4-benzonitrile formed hydrophobic interactions with Phe240, Met528 and Ile529. SAR studies119 have focused on the optimization of the side chain installed on the triazole pharmacophore to explore new antifungal agents in the last few years.
Fig. 24. The structures of isavuconazole (48), ravuconazole (50) and their derivatives.

Luliconazole (52) was initiatively developed by Niwano's group as a potent antifungal agent in 1998. Exhibiting a unique chemical structure that is characterized by incorporation of an imidazole group onto a ketene dithioacetal scaffold, this drug has a broad-spectrum antifungal activity against versatile dermatophytes including Trichophyton spp., Aspergillus spp., and Candida spp., which could cause superficial fungal infections affecting the hair, skin, and nail. Compared with another structure similar imidazole compound lanoconazole (53), which is prepared as a racemic mixture, the optically pure luliconazole (52) (only R isomer) has a much lower minimal inhibitory concentration against fungal species and therefore a better efficacy profile, which is mainly because their S enantiomers are both inactive (Fig. 25).120 In 2005, luliconazole (52) was firstly launched in Japan as a 1% cream followed by the US FDA approval in 2013 for treatment of tinea pedis, tinea cruris and tinea corporis caused by Trichophyton rubrum and Epidermophyton floccosum.121 Similar to isavuconazole (48) and ravuconazole (50), luliconazole (52) also shows its fungistatic property through inhibition of lanosterol 14α-demethylase and hence interrupting ergosterol biosynthesis. Molecular docking studies disclosed that this drug bound tightly to the cavity of lanosterol 14α-demethylase majorly via two important interactions. The first one was hydrogen bonding between the cyano group of the drug and residue Cys470 of the receptor, and the other was hydrophobic π–π stacking contact between the dichlorophenyl moiety with Tyr126.122 Due to its good penetration to α-keratin which is the main component of the nail bed, some investigations have been undertaken to apply luliconazole (52) in the treatment of onychomycosis that is a fungal infection affecting human nails recently.123,124
Fig. 25. The structures of luliconazole (52) and lanoconazole (53).

4.7. Antivirus drugs
Human immunodeficiency virus (HIV) is a well-known single-strand RNA virus causing acquired immunodeficiency syndrome (AIDS) by destroying the human immune system, especially CD4 positive T cells, eventually leading to death due to severe infection.125 Nowadays, combination antiretroviral therapy (CART) is the major method for the treatment of AIDS, which means that multiple (generally three) anti-HIV drugs with different targets are used together to treat AIDS in clinical practice to reduce the incidence of drug resistance and improve therapeutic effects.126 Generally, the first-line therapy contains two nucleoside reverse transcriptase inhibitors (NRTs) in combination with one drug belonging to other categories such as non-nucleoside reverse transcriptase inhibitors (NNRTIs), integrase strand transfer inhibitors (InSTIs), or protease inhibitors (PIs). Moreover, NNRTIs have been increasingly used in the CART regimen recently. For instance, complera, a combinational prescription127 approved in 2011, consists of rilpivirine (55) (NNRTI), emtricitabine (NRTI) and tenofovir (NRTI). Concerning the specific molecular mechanism of NNRTIs for anti-HIV activity, they bind to the allosteric site (NNRTI binding pocket, NNIBP) approximately in 10 Å distance from the polymerase active site of reverse transcriptase, thus resulting in a noncompetitive inhibition that blocks virus DNA synthesis through interfering with the interaction between the primer terminus and the polymerase active site.128 To date, there are six NNRTIs approved by the FDA that can be classified into first generation (nevirapine, delavirdine and efavirenz) and second generation (etravirine (54), rilpivirine (55) and doravirine (56)) (Fig. 26).129 Compared with the first generation NNRTIs, the newer ones have better efficacy especially in terms of reduced development of drug resistance as their flexible chemical structures can adapt more finely to the NNIBP through ‘wiggling and jiggling’.130 Among these second generation NNRTIs, etravirine (54) and rilpivirine (55), approved in 2008 and 2011 respectively, possess the same diarylpyrimidine scaffold as the basic pharmacophore.131 The utility of this skeleton as an NNRTI was first put forward by Tibotec and Janssen 30 years ago, which created a series of structurally related compounds exhibiting similar anti-HIV activities, and eventually etravirine (54) and rilpivirine (55) stood out from numerous candidates. These two drugs have a similar binding mode with reverse transcriptase, and rilpivirine (55) will be described thoroughly here. Molecular docking and crystallography showed that the left aromatic wing (cinnamonitrile moiety) is in contact with Y181, Y188, F227 and W229 via hydrophobic interaction, and the right one (benzonitrile moiety) formed interaction with V106, P225, P236 and Y318 in solvent exposed region I. Additionally, the nitrile group of the cinnamonitrile was reported132 to interact with water molecules resulting in improvement in binding affinity. Compared with etravirine (54), the cyanovinyl group was joined to the aryl by a longer linker that enhanced the flexibility of rilpivirine (55) and therefore increased its ability to combat more mutant resistance. However, this cyanovinyl group was more likely to undergo in vivo Michael addition with off-target substances to bring potential adverse events.133 As for doravirine (56), it is a novel chemical entity approved by the FDA in 2018, with a different chemical structure from the other two second-generation NNRTIs, thus leading to a different binding mode with reverse transcriptase. The crystal structure demonstrated that the benzonitrile part of doravirine had a π–π stacking interaction with the Y188 residue, and the linker between cyano and benzene was also shorter than rilpivirine (55), which may suggest that doravirine (56) had less flexibility to overcome some mutations.134
Fig. 26. The structures of non-nucleoside reverse transcriptase inhibitors.

In December 2019, a novel coronavirus named COVID-19 emerged around the world and rapidly became a pandemic disease within only 5 months. Up to now, the Word Health Organization has reported that there are over 130 million cases worldwide, with an estimated 2.87 million deaths due to severe respiratory disorders such as pneumonia caused by this virus. What is worse, these figures still steadily increase daily. Therefore, discovery of a potent therapy to treat this horrible infection is urgently needed and has attracted significant interest from big pharmaceutical companies. Remdesivir (57), a small molecule nucleoside analog, was indicated to be effective for the treatment of COVID-19 according to the latest clinical data.135 In terms of the molecular mechanism for its therapeutic potential, remdesivir is metabolized into its active form GS-441524 5′TS (58) after oral administration (Fig. 27). Then this active metabolite competes with endogenous nucleotides such as ATP for viral RNA-dependent RNA polymerase (RdRp) which is extremely important in the synthesis of viral RNA. Hence, remdesivir (57) is anticipated to combat COVID-19 via hampering RNA synthesis. Structure relationship analysis showed136,137 that the nitrile moiety in remdesivir (57) was accommodated in motif B of the active site and interacted with the S682 residue, which was regarded as a significant factor for the better antivirus activity of remdesivir (57) than that of other nucleoside analogue drugs. Additional study138 also disclosed that remdesivir (57) can exhibit antivirus properties via blockage of other potential targets involving vacuolar proton-translocating ATPase (V-ATPase) and angiotensin-converting enzyme 2 (ACE2).
Fig. 27. The structures of remdesivir (57) and its active metabolite (58).

5. Nitrile-containing drugs targeting central nervous system diseases
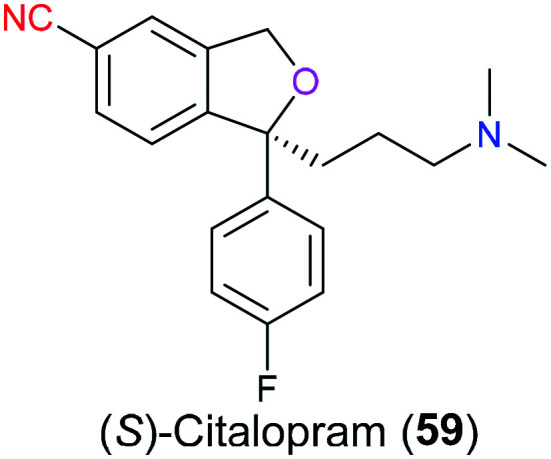
Serotonin (5-HT) is one of the most important neurotransmitters in our body that regulates various neurological activities related to the central nervous system such as sleep, pain and mood. Like most neurotransmitters, the synapse is the major site for serotonin to exert its function. More specifically, serotonin is released to the cleft after the fusion of the vesicle and presynaptic membrane, where it diffuses to bind to serotonin receptors located on the postsynaptic membrane. The activated receptors including several types of GPCR and ion channels then transduce the excitatory or inhibitory signal via a second messenger such as cAMP or membrane potential. Excess serotonin in the synaptic cleft will be recycled back to the presynaptic neuron through the serotonin reuptake receptor which is one of the neurotransmitter sodium symporters (NSSs). If there are some disorders on NSSs, several diseases associated with the nervous system such as depression may occur. Therefore, targeting the serotonin transporter is a promising strategy for the treatment of neurological disorders. (S)-Citalopram (59), also named escitalopram (Fig. 28), is a selective serotonin reuptake inhibitor that antagonizes serotonin transporters found on the presynaptic membrane. In 2016, Green and co-workers disclosed139 the X-ray structure and molecular mechanism for its binding behavior. When it refers to the contribution of the cyano group to the interaction between this drug and a serotonin transporter, a non-polar/polar surface formed between the nitrile moiety and V501 and F497 had to be emphasized. Moreover, the benzonitrile interacted with another aromatic ring in the pocket environment in an edge-to-face way rather than parallel stacking. Additionally, this study also suggested that (S)-citalopram (59) was more likely to exhibit better efficacy to T497A mutation than the wild type of the serotonin transporter as the former accommodated the cyano group better.
Fig. 28. The structure of (S)-citalopram (59).
Besides escitalopram, vilazodone (60) is another serotonergic antidepressant containing a nitrile group in its structure (Fig. 29), which was approved by the FDA in 2012 for treatment of major depressive disorder (MDD).140 Acting as a combined 5-HT1A receptor partial agonist and selective serotonin reuptake inhibitor, the onset of action of vilazodone is much faster than conventional SSRIs such as escitalopram. For conventional SSRIs, there is a therapeutic lag (at least 2 weeks) after drug intake due to the negative feedback mediated by presynaptic 5-HT1A autoreceptors, which counteracts the increased 5-HT induced by SSRIs. However, vilazodone (60) can promote desensitization of the 5-HT1A autoreceptor through its partial agonism activity and hence dramatically shorten the onset of action.141 SAR studies reported that the 5-cyano-indole-butyl-piperazine framework in this molecule exhibited SSRI activity and the rest of the scaffold (2-carboxamido benzofurane-5-yl group) was responsible for 5-HT1A receptor partial agonism.142 More specifically, the cyano group acted as a bioisostere of the fluorine atom and improved the affinity for blockade of the serotonin transporter owing to its electron-withdrawing property. Additionally, the flexible butyl linker between piperazine and indole also contributed to the enhanced affinity via displaying the best configuration in the active pocket.
Fig. 29. The structure of vilazodone (60).

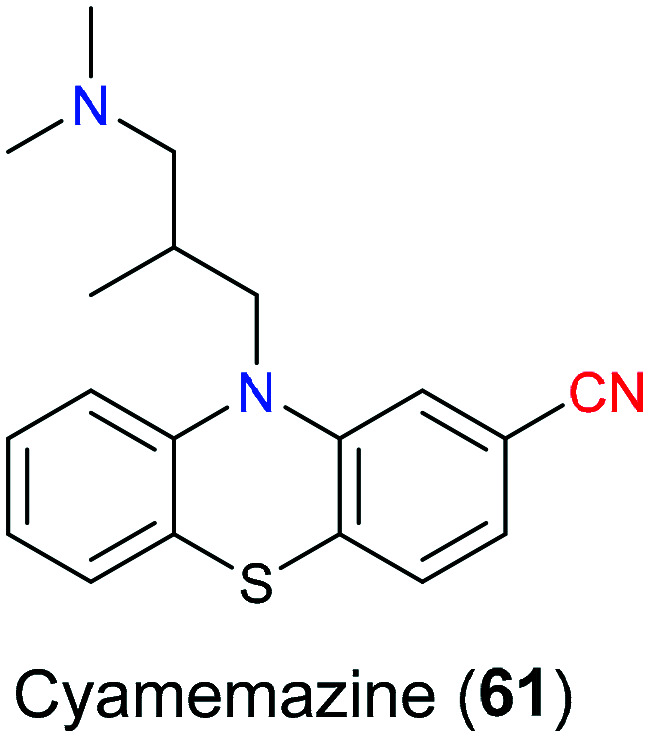
Cyamemazine (61), with a nitrile-containing phenothiazine scaffold (Fig. 30), is a neuroleptic antipsychotic used to treat schizophrenia and anxiety in clinical settings.143 It was reported144 that its neuroleptic activities were due to antagonism for D2 and D4 dopamine receptors, and its anxiolytic activity was ascribed to 5-HT3 and 5-HT2C antagonism.145 As for the detailed pharmacological action on the molecular level, there remained uncertainty.
Fig. 30. The structure of cyamemazine (61).
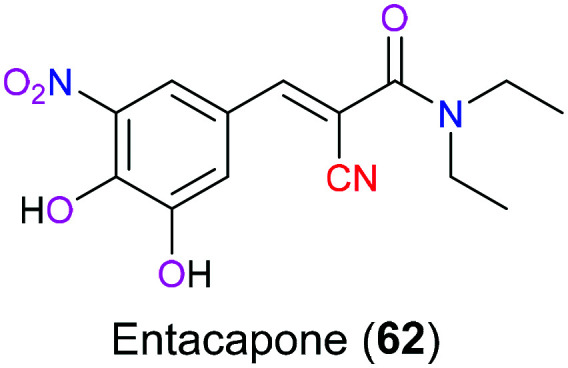
Entacapone (62) is a catechol-O-methyltransferase inhibitor (Fig. 31) that will effectively reduce the metabolism of catechol containing drugs such as levodopa which is a first-line therapy for the treatment Parkinson's syndrome. A study showed146 that administration of levodopa together with entacapone would enhance the therapeutic effects obviously due to the decreased metabolism and hence increased system exposure of levodopa.
Fig. 31. The structure of entacapone (62).
Ionotropic glutamate receptors (iGluRs) play an extremely significant role in excitatory neurotransmission which is closely related to central nervous system disorders including epileptogenesis as well as the generation and propagation of seizure. In particular, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) are generally considered to be the most vital subclass of iGluRs, which have aroused increasing interest of many medicinal chemists as a promising target for rational antiepileptic medicine design in recent years.147 The pyridine-based perampanel (63), a selective and noncompetitive AMPAR antagonist, was the first antiepileptic drug approved by the FDA in 2012 due to its much better safety and efficacy profile compared with other clinical candidates, although the high dose of this drug would lead to some adverse events such as dizziness and irritability.148 In 2016, Yelshanskaya et al. demonstrated149 the mechanism of noncompetitive inhibitory activity of perampanel (63) through molecular docking, which indicated that this drug as a negative allosteric mediator bound to the interface between the upper M3 transmembrane domain and S1-M1 and S2-M4 linkers connecting the transmembrane segment to the ligand-binding domain. Functioning as a wedge, perampanel (63) stabilized the closed conformation of AMPAR and interrupted gating rearrangement leading to ion channel opening. The nitrile group of this drug served as a hydrogen bond acceptor to interact with the Ser516 residue of the active site. SAR studies150 showed that the CN group position was able to determine the inhibitory efficacy of perampanel (63) derivatives. For example, its 4-CN isomer (65) exhibited an approximately 480-fold decrease in inhibitory property, and for its 3-cyano isomer (64), a 120-fold decrease could be observed. This was because the change of CN moiety position altered the geometry of aromatic rings leading to the hampered access of the 2-pyridyl group to the gating region (Fig. 32).
Fig. 32. The structure of perampanel (63) and other candidates.

Genetic alternations involving rearrangement and mutations have been reported to be associated with a broad variety of cancers. Taking lung cancer for example, the most common driver oncogene is Kirsten rat sarcoma viral oncogene (KRAS), followed by epidermal growth factor receptor gene (EGFR) and anaplastic lymphoma kinase gene (ALK).151 In recent years, ALK has become increasingly important as a promising target for the treatment of ALK-positive non-small cell lung cancer (NSCLC), which accounts for approximately 3–7% of advanced NSCLC.152 ALK is a transmembrane glycoprotein belonging to the insulin receptor subfamily of receptor tyrosine kinases and its structure includes an extracellular ligand-binding domain, a transmembrane domain, and cytoplasmic tyrosine kinase domain. Chromosomal translocation resulting in a fusion of ALK with echinoderm microtubule-associated protein-like 4 (EML4) will produce a chimeric tyrosine kinase protein with a constitutive activity, which ultimately causes loss-of-control cell proliferation and therefore tumor growth via a series of sharply increased phosphorylation-dependent signal transductions.153 Versatile ALK inhibitors have been approved by the FDA for the treatment of ALK-driven NSCLC in the last few decades due to gradually growing drug resistance, including alectinib (66) (a second-generation ALK inhibitor approved in 2015) and lorlatinib (67) (a third-generation inhibitor approved in 2018). Owing to the superior medicinal properties of alectinib (66), the FDA had awarded this drug with the title “Breakthrough Therapy Designation” (Fig. 33). In 2011, Sakamoto et al. analyzed154 the X-ray crystal structure of the binding pattern of alectinib (66) with the ALK protein and disclosed the specific interaction between the chemical structure of alectinib (66) and the active site of this target via molecular dynamics simulation. The model suggested that alectinib (66) was located in the sandwich pocket (ATP binding site) between the N- and C-lobe, with the substituted nitrile group of the unique benzo[b]carbazole scaffold pointed to the αC-helix.
Fig. 33. The structures of alectinib (66) and lorlatinib (67).

This nitrile moiety mainly participated in the formation of hydrogen bond interaction with nearby amino acid residues Lys1150, Arg1253, Gly1269 and Glu1270 with the aid of water molecules. Moreover, the carbon atom of this carbonitrile group was involved in the CH/π interaction with the neighboring Leu1196 of the naive ALK protein. Even in the L1196M mutant model, this interaction also existed with the mutated residue Met, which exactly explained why alectinib (66) could effectively overcome the resistance to crizotinib with L1196M mutation.155 As for lorlatinib (67), it has a broader therapeutical activity than alectinib (66) because it can overcome 1151T-ins and G1202R ALK mutations that make alectinib (66) useless.156 The macrocycle is the most appealing feature in the molecular structure of lorlatinib (67), which is thought to contribute to the enhanced chemical and metabolic stability and reduce the possibility of P-glycoprotein 1 mediated efflux.157 As for the role of its pyrazole nitrile scaffold, molecular docking analysis disclosed that there were unfavorable interactions between the nitrile group and the adjacent carbon atom of the residue Tyr635 in TrkB, which allow this nitrile group to dramatically improve the binding selectivity of this drug. SAR studies demonstrated that if it was replaced with a methyl group, a less than 35-fold selectivity was shown. Additionally, electrostatics as a consequence of the proximity of the electron donor nitrogen atom of the cyano group and tyrosine could be another important contributor to the improvement of the selectivity.158
Selpercatinib (68) was approved by the FDA in 2020 targeting the rearranged during transfection (RET) gene that encodes the receptor tyrosine kinase (Fig. 34). As a tyrosine kinase inhibitor (TKI), selpercatinib (68) was reported to show a remarkable safety profile and anticancer activity in a broad variety of diseases including RET-fusion positive non-small cell lung cancer (NSCLC), advanced or metastatic RET-mutant medullary thyroid cancer (MTC) and differentiated thyroid carcinoma. Not only wild-type RET but also diverse mutated RET isoforms could be effectively inhibited by this drug.159,160 The X-ray crystal structure of the RET kinase–selpercatinib complex disclosed the formation of a hydrogen bond between the side chain of A807 and a pyrazolo nitrogen from this pharmaceutical molecule. Additionally, this drug also hydrophobically interacted with Y806 via the pyrazolo[1,5-α]pyridine ring and the hydroxymethyl group, which was located in the adenosine pocket.161 As for the pyridine ring of this drug, it occupied the front pocket-I and was in contact with V738 of the β2 strand through van der Waals interactions.162 However, the function of the nitrile group in this drug molecule has not been illustrated.
Fig. 34. The structure of selpercatinib (68).

6. Nitrile-containing drugs targeting gastrointestinal tract diseases
Gastric ulcer is one of the most frequently occurring diseases related to the gastrointestinal tract (GI tract), accounting for 4% in the population. Histamine is an aminergic endogenous substance secreted by immune cells including basophils and mast cells. Its immunomodulatory property can lead to stomach ulcer by binding to histamine receptor 2 (H2R).
More specifically, once the extracellular domain of H2R located on gastric parietal cells receives histamine, its conformation is changed to activate the coupled G protein, and then signals will be transduced to adenylyl cyclase which can elevate the cellular level of second messenger cAMP. Increased cAMP will activate protein kinase A which induces a cascade of biochemical sequences including enhanced/blocked key enzyme activity and/or gene expression via phosphorylation of certain amino acid residues such as serine and tyrosine. Ultimately, gastric acid secretion of stomach cells will be increased, which can aggravate ulcer condition.163 Hence, targeting H2R is a potential strategy for the treatment of gastric ulcer. Cimetidine (69) is one of the H2R antagonists that block the signalling pathway mediated by H2R, approved by the FDA to relieve peptic ulcer in 1979.164 Molecular docking showed that the H2R antagonism activity of cimetidine (69) relied on the hydrogen bond interactions of its imidazole part with the D98 residue at transmembrane helices III of H2R. Additionally, Y250 at transmembrane helices VI is in contact with cimetidine (69) via hydrogen bonding and hydrophobic interaction. In terms of the role of the cyano moiety in cimetidine (69), it was reported that this group formed a hydrogen bond with T190. Moreover, owing to the cyanoguanidine fragment of cimetidine (69), this drug had slightly better pharmacological effects than the structure-related drug metiamide (70) (Fig. 35).
Fig. 35. The structures of cimetidine (69) and metiamide (70).

7. Nitrile-containing drugs as dietary supplements
Vitamin B12 (71) is one of the naturally occurring products that contain the nitrile group (Fig. 36). Humans cannot synthesize this vital compound by themselves and we can only acquire it from dietary food, for example, beef and animal liver. Old or strict vegetarians easily suffer from vitamin B12 deficiency characterized by megaloblastic anemia and impaired neurological function.165 In terms of its chemical structure, the cobalt(iii) ion is centered in the amidated tetrapyrrole macrocycle to form coordination contact with four pyrrole nitrogen atoms and one dimethylbenzimidazole nitrogen atom. As there is one cyano group that interacts covalently with this cobalt, vitamin B12 (71) is also known as cyanocobalamin (CNCbl) which is usually present in the synthetic industry in fortified foods or vitamin pills to treat vitamin B12 deficiency. After absorption of vitamin B12 (71) into the circulatory system, the cyano group will be substituted by a variety of moieties including the adenosyl group, methyl group, and hydroxyl group to generate AdoCbl, MeCbl and OHCbl, respectively. These types of vitamin B12 (71) display a particularly significant role in nucleic acid synthesis, neurological function and blood cell formation as coenzymes for certain crucial biochemical processes such as methylation, isomerization, radical S-adenosylmethionine and reductive dehalogenation. Taking 5-deoxy AdoCbl as an example, it acts as a cofactor of methylmalonyl CoA mutase to help the formation of succinyl CoA from methylmalonyl CoA, which is an extremely vital process for the normal function of the nervous system.166 There is a study showing that the introduction of the cyano group in vitamin B12 (71) makes it more chemically stable for manufacture compared with its native form (OHCbl). Although CNCbl will give a higher plasma concentration of vitamin B12 (71) than OHCbl, no additional benefits result from this, which is because the extra CNCbl binds to haptocorrin as an inactive form. Furthermore, evidence shows that the kidney is the main place for CBCbl accumulation, while OHCbl favored the liver.167
Fig. 36. The structure of vitamin B12 (71).

Conclusions
Although the application of nitrile units in structure-based drug design is insufficient compared with traditional strategies including methyl substitution, the introduction of this functional group into drug molecules has increasingly aroused great interest from medicinal chemists due to its regulatory properties for the pharmacokinetic profile and binding mode. For example, nitrile moieties, especially α,β-unsaturated nitriles, are easily attacked by internal nucleophiles such as the hydroxyl group of the serine residue to afford an adduct reversibly or irreversibly, which provides a new insight for medicinal chemists to design novel and potent small molecule inhibitors exhibiting anticancer or antiviral activities in the future. Moreover, displacement of aromatic nitrogen, carbonyl, halogen, methyl, and other bioisosteres with nitrile substituents is less investigated for a variety of lead compounds in SAR studies. However, with an in-depth understanding of the interaction between the nitrile group and the receptor protein, more and more nitrile-containing drug molecules via bioisostere substitution are expected to be developed. Furthermore, the nitrile substitution strategy can also be widely used to explore next-generation pharmaceuticals with less drug resistance in the coming days. We expect that this review could bring some inspiration to both organic and medicinal chemists in drug discovery with chemical entities bearing nitrile groups.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We acknowledge the National Natural Science Foundation of China (#22071175) for financial support, and we also thank Prof. Niu Huang [National Institute of Biological Sciences, Beijing] for helpful suggestions and comments.
References
- Singh P. D. Jackson J. R. James S. P. Biochem. Pharmacol. 1985;34:2207–2209. doi: 10.1016/0006-2952(85)90420-4. [DOI] [PubMed] [Google Scholar]
- Wang Y. Du Y. Huang N. Future Med. Chem. 2018;10:2713–2727. doi: 10.4155/fmc-2018-0252. [DOI] [PubMed] [Google Scholar]
- Nabeno M. Akahoshi F. Kishida H. Miyaguchi I. Tanaka Y. Ishii S. Kadowaki T. Biochem. Biophys. Res. Commun. 2013;434:191–196. doi: 10.1016/j.bbrc.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Fleming F. F. Yao L. Ravikumar P. C. Funk L. Shook B. C. J. Med. Chem. 2010;53:7902–7917. doi: 10.1021/jm100762r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming F. F. Nat. Prod. Rep. 1999;16:597–606. doi: 10.1039/A804370A. [DOI] [Google Scholar]
- Wang J. Liu H. Chin. J. Org. Chem. 2012;32:1643–1652. doi: 10.6023/cjoc1202132. [DOI] [Google Scholar]
- Foster M. N. Coetzee W. A. Physiol. Rev. 2016;96:177–252. doi: 10.1152/physrev.00003.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S. Wang M. Ma Z. Brain Res. Bull. 2021;169:1–7. doi: 10.1016/j.brainresbull.2021.01.003. [DOI] [PubMed] [Google Scholar]
- Kaiser J. Krippeit-Drews P. Drews G. Diabetes. 2021;70:423–435. doi: 10.2337/db20-0231. [DOI] [PubMed] [Google Scholar]
- Salamon E. Mannhold R. Weber H. Lemoine H. Frank W. J. Med. Chem. 2002;45:1086–1097. doi: 10.1021/jm010999g. [DOI] [PubMed] [Google Scholar]
- Horino H. Mimura T. Kagechika K. Ohta M. Kubo H. Kitagawa M. Chem. Pharm. Bull. 1998;46:602–609. doi: 10.1248/cpb.46.602. [DOI] [PubMed] [Google Scholar]
- Munoz-Islas E. Gupta S. Jimenez-Mena L. R. Lozano-Cuenca J. Sanchez-Lopez A. Centurion D. Mehrotra S. MaassenVanDenBrink A. Villalon C. M. Br. J. Pharmacol. 2006;149:82–91. doi: 10.1038/sj.bjp.0706839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Nafria J. Nehme R. Edwards P. C. Tate C. G. Nature. 2018;558:620–623. doi: 10.1038/s41586-018-0241-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonov D. B. Lin L. Yang D. S. C. Yuchi Z. Zhorov B. S. J. Comput.-Aided Mol. Des. 2020;34:1157–1169. doi: 10.1007/s10822-020-00330-0. [DOI] [PubMed] [Google Scholar]
- Cheng R. C. K. Tikhonov D. B. Zhorov B. S. J. Biol. Chem. 2009;284:28332–28342. doi: 10.1074/jbc.M109.027326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster G. Bekema H. J. Wetterslev J. Gluud C. Keus F. van der Horst I. C. C. Intensive Care Med. 2016;42:1322–1335. doi: 10.1007/s00134-016-4449-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H. S. Joo J.-D. Kim D.-W. In J. H. Roh M. Jeong J.-T. Noh S. J. Choi J. W. Korean J. Anesthesiol. 2014;66:136–142. doi: 10.4097/kjae.2014.66.2.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uysal E. Dokur M. Altinay S. Saygili E. I. Batcioglu K. Ceylan M. S. Kazimoglu H. Uyumlu B. A. Karadag M. J. Invest. Surg. 2018;31:402–411. doi: 10.1080/08941939.2017.1343880. [DOI] [PubMed] [Google Scholar]
- Saklani R. Jaggi A. Singh N. Arch. Pharmacal Res. 2010;33:1049–1057. doi: 10.1007/s12272-010-0711-6. [DOI] [PubMed] [Google Scholar]
- Rao V. Ivanov J. Weisel R. D. Ikonomidis J. S. Christakis G. T. David T. E. J. Thorac. Cardiovasc. Surg. 1996;112:38–51. doi: 10.1016/S0022-5223(96)70176-9. [DOI] [PubMed] [Google Scholar]
- Dorigo P. Fraccarollo D. Santostasi G. Gaion R. M. Maragno I. Floreani M. Carpenedo F. Jester M. Mosti L. Schenone P. Farmaco. 1994;49:19–23. [PubMed] [Google Scholar]
- Cicalini I. De Filippis B. Gambacorta N. Di Michele A. Valentinuzzi S. Ammazzalorso A. Della Valle A. Amoroso R. Nicolotti O. Del Boccio P. Giampietro L. Molecules. 2020;25:1817–1830. doi: 10.3390/molecules25081817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan A. P. Allergy Asthma Proc. 2020;41:S51–S54. doi: 10.2500/aap.2020.41.200056. [DOI] [PubMed] [Google Scholar]
- Wedner H. J. Allergy Asthma Proc. 2020;41:S14–S17. doi: 10.2500/aap.2020.41.200081. [DOI] [PubMed] [Google Scholar]
- Zuraw B. L. Christiansen S. C. Clin. Rev. Allergy Immunol. 2016;51:216–229. doi: 10.1007/s12016-016-8561-8. [DOI] [PubMed] [Google Scholar]
- Ohsawa I. Honda D. Suzuki Y. Fukuda T. Kohga K. Morita E. Moriwaki S. Ishikawa O. Sasaki Y. Tago M. Chittick G. Cornpropst M. Murray S. C. Dobo S. M. Nagy E. Van Dyke S. Reese L. Best J. M. Iocca H. Collis P. Sheridan W. P. Hide M. Allergy. 2020:1–11. doi: 10.1111/all.14670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J. R. Hwang G. Johri A. Craig T. Immunotherapy. 2019;11:1439–1444. doi: 10.2217/imt-2019-0128. [DOI] [PubMed] [Google Scholar]
- Rajaram P. Rivera A. Muthima K. Olveda N. Muchalski H. Chen Q.-H. Molecules. 2020;25:2448–2474. doi: 10.3390/molecules25102448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duke, III C. B. Jones A. Bohl C. E. Dalton J. T. Miller D. D. J. Med. Chem. 2011;54:3973–3976. doi: 10.1021/jm2000097. [DOI] [PubMed] [Google Scholar]
- Boukovala M. Spetsieris N. Efstathiou E. Expert Opin. Pharmacother. 2020;21:1537–1546. doi: 10.1080/14656566.2020.1770726. [DOI] [PubMed] [Google Scholar]
- Yu J. Zhou P. Hu M. Yang L. Yan G. Xu R. Deng Y. Li X. Chen Y. Eur. J. Med. Chem. 2019;182:111608–111627. doi: 10.1016/j.ejmech.2019.111608. [DOI] [PubMed] [Google Scholar]
- Tucker H. Crook J. W. Chesterson G. J. J. Med. Chem. 1988;31:954–959. doi: 10.1021/jm00400a011. [DOI] [PubMed] [Google Scholar]
- Pertusati F. Ferla S. Bassetto M. Brancale A. Khandil S. Westwell A. D. McGuigan C. Eur. J. Med. Chem. 2019;180:1–14. doi: 10.1016/j.ejmech.2019.07.001. [DOI] [PubMed] [Google Scholar]
- Mojaddami A. Sakhteman A. Fereidoonnezhad M. Faghih Z. Najdian A. Khabnadideh S. Sadeghpour H. Rezaei Z. Res. Pharm. Sci. 2017;12:21–30. doi: 10.4103/1735-5362.199043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Otaibi J. S. Almuqrin A. H. Mary Y. S. Mary Y. S. J. Mol. Struct. 2020;1217:128388–128394. doi: 10.1016/j.molstruc.2020.128388. [DOI] [Google Scholar]
- Adhikari N. Amin S. A. Saha A. Jha T. Eur. J. Med. Chem. 2017;137:365–438. doi: 10.1016/j.ejmech.2017.05.041. [DOI] [PubMed] [Google Scholar]
- Buzdar A. U. Robertson J. F. R. Eiermann W. Nabholtz J. M. Cancer. 2002;95:2006–2016. doi: 10.1002/cncr.10908. [DOI] [PubMed] [Google Scholar]
- Hynes N. E. Lane H. A. Nat. Rev. Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Yarden Y. Sliwkowski M. X. Nat. Rev. Mol. Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Cho H. S. Mason K. Ramyar K. X. Stanley A. M. Gabelli S. B. Denney D. W. Leahy D. J. Nature. 2003;421:756–760. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- Baselga J. Swain S. M. Nat. Rev. Cancer. 2009;9:463–475. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- Bose P. Ozer H. Expert Opin. Invest. Drugs. 2009;18:1735–1751. doi: 10.1517/13543780903305428. [DOI] [PubMed] [Google Scholar]
- Rabindran S. K. Discafani C. M. Rosfjord E. C. Baxter M. Floyd M. B. Golas J. Hallett W. A. Johnson B. D. Nilakantan R. Overbeek E. Reich M. F. Shen R. Shi X. Q. Tsou H. R. Wang Y. F. Wissner A. Cancer Res. 2004;64:3958–3965. doi: 10.1158/0008-5472.CAN-03-2868. [DOI] [PubMed] [Google Scholar]
- Wissner A. Overbeek E. Reich M. F. Floyd M. B. Johnson B. D. Mamuya N. Rosfjord E. C. Discafani C. Davis R. Shi X. Q. Rabindran S. K. Gruber B. C. Ye F. Hallett W. A. Nilakantan R. Shen R. Wang Y. F. Greenberger L. M. Tsou H. R. J. Med. Chem. 2003;46:49–63. doi: 10.1021/jm020241c. [DOI] [PubMed] [Google Scholar]
- Wissner A. Floyd M. B. Rabindran S. K. Nilakantan R. Greenberger L. M. Shen R. Wang Y. F. Tsou H. R. Bioorg. Med. Chem. Lett. 2002;12:2893–2897. doi: 10.1016/S0960-894X(02)00598-X. [DOI] [PubMed] [Google Scholar]
- Wen H.-N. Liu Y.-X. Xu D. Zhao K.-J. Jiao Z. Eur. J. Pharm. Sci. 2021;159:105729–105736. doi: 10.1016/j.ejps.2021.105729. [DOI] [PubMed] [Google Scholar]
- Barbot M. Zilio M. Scaroni C. Best Pract. Res. Clin. Endocrinol. Metab. 2020;34:101380–101402. doi: 10.1016/j.beem.2020.101380. [DOI] [PubMed] [Google Scholar]
- Lonser R. R. Nieman L. Oldfield E. H. J. Neurosurg. 2017;126:404–417. doi: 10.3171/2016.1.JNS152119. [DOI] [PubMed] [Google Scholar]
- Duggan S. Drugs. 2020;80:495–500. doi: 10.1007/s40265-020-01277-0. [DOI] [PubMed] [Google Scholar]
- Usanov S. A. Kliuchenovich A. V. Strushkevich N. V. Expert Opin. Drug Discovery. 2019;14:143–151. doi: 10.1080/17460441.2019.1559146. [DOI] [PubMed] [Google Scholar]
- Sachinidis A. Stoica R. A. Nikolic D. Patti A. M. Rizvi A. A. Metabolism. 2020;109:154295. doi: 10.1016/j.metabol.2020.154295. [DOI] [PubMed] [Google Scholar]
- Wicinski M. Gorski K. Wodkiewicz E. Walczak M. Nowaczewska M. Malinowski B. Int. J. Mol. Sci. 2020;21:2275–2291. doi: 10.3390/ijms21072275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J.-U. Curr. Top. Med. Chem. 2007;7:579–595. doi: 10.2174/156802607780091000. [DOI] [PubMed] [Google Scholar]
- Gupta R. Walunj S. S. Tokala R. K. Parsa K. V. L. Singh S. K. Pal M. Curr. Drug Targets. 2009;10:71–87. doi: 10.2174/138945009787122860. [DOI] [PubMed] [Google Scholar]
- Hochhaus A. La Rosee P. Leukemia. 2004;18:1321–1331. doi: 10.1038/sj.leu.2403426. [DOI] [PubMed] [Google Scholar]
- Lu T. Cao J. Zou F. Li X. Wang A. Wang W. Liang H. Liu Q. Hu C. Chen C. Hu Z. Wang W. Li L. Ge J. Shen Y. Ren T. Liu J. Xia R. Liu Q. Eur. J. Pharmacol. 2021;897:173944–173951. doi: 10.1016/j.ejphar.2021.173944. [DOI] [PubMed] [Google Scholar]
- Roskoski, Jr. R. Pharmacol. Res. 2015;94:9–25. doi: 10.1016/j.phrs.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Singh A. N. Sharma N. Protein J. 2019;38:537–550. doi: 10.1007/s10930-019-09832-9. [DOI] [PubMed] [Google Scholar]
- Munchhof M. J. Li Q. Shavnya A. Borzillo G. V. Boyden T. L. Jones C. S. LaGreca S. D. Martinez-Alsina L. Patel N. Pelletier K. Reiter L. A. Robbins M. D. Tkalcevic G. T. ACS Med. Chem. Lett. 2012;3:106–111. doi: 10.1021/ml2002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L. White D. W. Gross S. Bennett B. D. Bittinger M. A. Driggers E. M. Fantin V. R. Jang H. G. Jin S. Keenan M. C. Marks K. M. Prins R. M. Ward P. S. Yen K. E. Liau L. M. Rabinowitz J. D. Cantley L. C. Thompson C. B. Vander Heiden M. G. Su S. M. Nature. 2010;465:966–966. doi: 10.1038/nature09132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNardo C. D. Ravandi F. Agresta S. Konopleva M. Takahashi K. Kadia T. Routbort M. Patel K. P. Brandt M. Pierce S. Garcia-Manero G. Cortes J. Kantarjian H. Am. J. Hematol. 2015;90:732–736. doi: 10.1002/ajh.24072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassereddine S. Lap C. J. Tabbara I. A. OncoTargets Ther. 2019;12:303–308. doi: 10.2147/OTT.S182443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe S. Wang H. DiNardo C. D. Stein E. M. de Botton S. Roboz G. J. Altman J. K. Mims A. S. Watts J. M. Pollyea D. A. Fathi A. T. Tallman M. S. Kantarjian H. M. Stone R. M. Quek L. Konteatis Z. Dang L. Nicolay B. Nejad P. Liu G. Zhang V. Liu H. Goldwasser M. Liu W. Marks K. Bowden C. Biller S. A. Attar E. C. Wu B. Blood Adv. 2020;4:1894–1905. doi: 10.1182/bloodadvances.2020001503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovici-Muller J. Lemieux R. M. Artin E. Saunders J. O. Salituro F. G. Travins J. Cianchetta G. Cai Z. Zhou D. Cui D. Chen P. Straley K. Tobin E. Wang F. David M. D. Penard-Lacronique V. Quivoron C. Saada V. De Botton S. Gross S. Dang L. Yang H. Utley L. Chen Y. Kim H. Jin S. Gu Z. Yao G. Luo Z. Lv X. Fang C. Yan L. Olaharski A. Silverman L. Biller S. Su S.-S. M. Yen K. ACS Med. Chem. Lett. 2018;9:300–305. doi: 10.1021/acsmedchemlett.7b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fostea R. M. Fontana E. Torga G. Arkenau H.-T. Cancers. 2020;12:2599–2610. doi: 10.3390/cancers12092599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Salama Z. T. Scott L. J. Drugs. 2018;78:761–772. doi: 10.1007/s40265-018-0908-4. [DOI] [PubMed] [Google Scholar]
- Kuriya B. Cohen M. D. Keystone E. Ther. Adv. Musculoskeletal Dis. 2017;9:37–44. doi: 10.1177/1759720X16687481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertsias G. Mediterr. J. Rheumatol. 2020;31:105–111. doi: 10.31138/mjr.31.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagca B. G. Avci C. B. Cytokine Growth Factor Rev. 2020;54:51–61. doi: 10.1016/j.cytogfr.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R. R. Li B. Yun S. Y. Chan A. Nareddy P. Gunawan S. Ayaz M. Lawrence H. R. Reuther G. W. Lawrence N. J. Schonbrunn E. J. Med. Chem. 2021;64:2228–2241. doi: 10.1021/acs.jmedchem.0c01952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samjoo I. A. Worthington E. Drudge C. Zhao M. Cameron C. Haring D. A. Stoneman D. Klotz L. Adlard N. J. Comp. Eff. Res. 2021;10:495–507. doi: 10.2217/cer-2020-0267. [DOI] [PubMed] [Google Scholar]
- Swallow E. Patterson-Lomba O. Yin L. Mehta R. Pelletier C. Kao D. Sheffield J. K. Stonehouse T. Signorovitch J. J. Comp. Eff. Res. 2020;9:275–286. doi: 10.2217/cer-2019-0169. [DOI] [PubMed] [Google Scholar]
- Sun Y. Yang Y. Wang Z. Jiang F. Chen Z. Wang Z. Front. Pharmacol. 2020;11:589146. doi: 10.3389/fphar.2020.589146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J. Giovannoni G. Hunter S. F. Drugs. 2021;81:207–231. doi: 10.1007/s40265-020-01431-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Zaqri N. Pooventhiran T. Rao D. J. Alsalme A. Warad I. Thomas R. J. Mol. Liq. 2020;318:114082. doi: 10.1016/j.molliq.2020.114082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Costa Nunes I. K. de Souza E. T. Roseno Martins I. R. Barbosa G. de Moraes Junior, Jr. M. O. Medeiros M. D. M. Silva S. W. Duarte. Balliano T. L. da Silva B. A. Silva P. M. Rodrigues. Carvalho V. D. F. Martins M. A. Lima L. M. Eur. J. Med. Chem. 2020;204:112492. doi: 10.1016/j.ejmech.2020.112492. [DOI] [PubMed] [Google Scholar]
- Liu F.-C. Yu H.-P. Lin C.-Y. Elzoghby A. O. Hwang T.-L. Fang J.-Y. J. Nanobiotechnol. 2018;16:35. doi: 10.1186/s12951-018-0364-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Card G. L. England B. P. Suzuki Y. Fong D. Powell B. Lee B. Luu C. Tabrizizad M. Gillette S. Ibrahim P. N. Artis D. R. Bollag G. Milburn M. V. Kim S. H. Schlessinger J. Zhang K. Y. J. Structure. 2004;12:2233–2247. doi: 10.1016/j.str.2004.10.004. [DOI] [PubMed] [Google Scholar]
- El-Husseiny W. M. El-Sayed M. A. A. El-Azab A. S. AlSaif N. A. Alanazi M. M. Abdel-Aziz A. A. M. J. Enzyme Inhib. Med. Chem. 2020;35:744–758. doi: 10.1080/14756366.2020.1740695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breedveld F. C. Dayer J. M. Ann. Rheum. Dis. 2000;59:841–849. doi: 10.1136/ard.59.11.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann P. Mandl-Weber S. Voelkl A. Adam C. Bumeder I. Oduncu F. Schmidmaier R. Mol. Cancer Ther. 2009;8:366–375. doi: 10.1158/1535-7163.MCT-08-0664. [DOI] [PubMed] [Google Scholar]
- Zhu S. Yan X. Xiang Z. Ding H.-F. Cui H. PLoS One. 2013;8:e71555. doi: 10.1371/journal.pone.0071555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalgutkar A. S. Nguyen H. T. Vaz A. D. N. Doan A. Dalvie D. K. Drug Metab. Dispos. 2003;31:1240–1250. doi: 10.1124/dmd.31.10.1240. [DOI] [PubMed] [Google Scholar]
- Klotz L. Eschborn M. Lindner M. Liebmann M. Herold M. Janoschka C. Garrido B. T. Schulte-Mecklenbeck A. Gross C. C. Breuer J. Hundehege P. Posevitz V. Pignolet B. Nebel G. Glander S. Freise N. Austermann J. Wirth T. Campbell G. R. Schneider-Hohendorf T. Eveslage M. Brassat D. Schwab N. Loser K. Roth J. Busch K. B. Stoll M. Mahad D. J. Meuth S. G. Turner T. Bar-Or A. Wiendl H. Sci. Transl. Med. 2019;11:eaao5563. doi: 10.1126/scitranslmed.aao5563. [DOI] [PubMed] [Google Scholar]
- Fragoso Y. D. Bidin Brooks J. B. Expert Rev. Clin. Pharmacol. 2015;8:315–320. doi: 10.1586/17512433.2015.1019343. [DOI] [PubMed] [Google Scholar]
- Liu S. P. Neidhardt E. A. Grossman T. H. Ocain T. Clardy J. Structure. 2000;8:25–33. doi: 10.1016/S0969-2126(00)00077-0. [DOI] [PubMed] [Google Scholar]
- Panek J. J. Jezierska A. Mierzwicki K. Latajka Z. Koll A. J. Chem. Inf. Model. 2005;45:39–48. doi: 10.1021/ci049754d. [DOI] [PubMed] [Google Scholar]
- Shekelle P. G. Newberry S. J. Ann. Intern. Med. 2017;166:855. doi: 10.7326/L17-0212. [DOI] [PubMed] [Google Scholar]
- Cuenca J. A. Balda J. Palacio A. Young L. Pillinger M. H. Tamariz L. Int. J. Rheumatol. 2019;2019:1076189. doi: 10.1155/2019/1076189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K.-H. Recent Pat. Inflammation Allergy Drug Discovery. 2007;1:69–75. doi: 10.2174/187221307779815020. [DOI] [PubMed] [Google Scholar]
- Hille R. Chem. Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- Okamoto K. Matsumoto K. Hille R. Eger B. T. Pai E. F. Nishino T. Proc. Natl. Acad. Sci. U. S. A. 2004;101:7931–7936. doi: 10.1073/pnas.0400973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K. Eger B. T. Nishino T. Kondo S. Pai E. F. Nishino T. J. Biol. Chem. 2003;278:1848–1855. doi: 10.1074/jbc.M208307200. [DOI] [PubMed] [Google Scholar]
- Matsumoto K. Okamoto K. Ashizawa N. Nishino T. J. Pharmacol. Exp. Ther. 2011;336:95–103. doi: 10.1124/jpet.110.174540. [DOI] [PubMed] [Google Scholar]
- McCormack P. L. Drugs. 2010;70:2073–2088. doi: 10.2165/11206320-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Ruan X. Seeger H. Mueck A. O. Maturitas. 2012;71:337–344. doi: 10.1016/j.maturitas.2012.01.018. [DOI] [PubMed] [Google Scholar]
- Torres T. Acta Med. Port. 2017;30:669–670. doi: 10.20344/amp.9787. [DOI] [PubMed] [Google Scholar]
- Fahrbach K. Tarpey J. Washington E. B. Hughes R. Thom H. Neary M. P. Cha A. Gerber R. Cappelleri J. C. Dermatol. Ther. 2020;10:1441–1444. doi: 10.1007/s13555-020-00452-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. Wang J. Zhang X. JAMA Dermatol. 2019;155:865–865. doi: 10.1001/jamadermatol.2019.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund Y. R. Akama T. Alley M. R. K. Antunes J. Dong C. Jarnagin K. Kimura R. Nieman J. A. Maples K. R. Plattner J. J. Rock F. Sharma R. Singh R. Sanders V. Zhou Y. FEBS Lett. 2012;586:3410–3414. doi: 10.1016/j.febslet.2012.07.058. [DOI] [PubMed] [Google Scholar]
- Akama T. Baker S. J. Zhang Y.-K. Hernandez V. Zhou H. Sanders V. Freund Y. Kimura R. Maples K. R. Plattner J. J. Bioorg. Med. Chem. Lett. 2009;19:2129–2132. doi: 10.1016/j.bmcl.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Chu Z. Xu Q. Zhu Q. Ma X. Mo J. Lin G. Zhao Y. Gu Y. Bian L. Shao L. Guo J. Ye W. Li J. He G. Xu Y. Eur. J. Med. Chem. 2021;213:113171–113171. doi: 10.1016/j.ejmech.2021.113171. [DOI] [PubMed] [Google Scholar]
- Sassetti E. Cruz C. D. Tammela P. Winterhalter M. Augustyns K. Gribbon P. Windshuegel B. Int. J. Mol. Sci. 2019;20:2686. doi: 10.3390/ijms20112686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeage K. Drugs. 2015;75:687–693. doi: 10.1007/s40265-015-0384-z. [DOI] [PubMed] [Google Scholar]
- Hogan C. A. Puri L. Gore G. Pai M. J. Clin. Tuberc. Other Mycobact. Dis. 2017;6:1–7. doi: 10.1016/j.jctube.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J. L. Grinius L. Hooper D. C. J. Bacteriol. 2002;184:1370–1377. doi: 10.1128/JB.184.5.1370-1377.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoletti R. Cai T. Perletti G. Wagenlehner F. M. E. Johansen T. E. B. Expert Opin. Invest. Drugs. 2015;24:957–963. doi: 10.1517/13543784.2015.1052401. [DOI] [PubMed] [Google Scholar]
- Peterson L. R. Clin. Infect. Dis. 2001;33:S180–S186. doi: 10.1086/321846. [DOI] [PubMed] [Google Scholar]
- Emami S. Shafiee A. Foroumadi A. Mini-Rev. Med. Chem. 2006;6:375–386. doi: 10.2174/138955706776361493. [DOI] [PubMed] [Google Scholar]
- Suaifan G. A. R. Y. Mohammed A. A. M. Bioorg. Med. Chem. 2019;27:3005–3060. doi: 10.1016/j.bmc.2019.05.038. [DOI] [PubMed] [Google Scholar]
- Silley P. Stephan B. Greife H. A. Pridmore A. Vet. Microbiol. 2012;157:106–111. doi: 10.1016/j.vetmic.2011.11.027. [DOI] [PubMed] [Google Scholar]
- Koehler P. Comely O. A. Infect. Dis. Clin. North Am. 2016;30:265–275. doi: 10.1016/j.idc.2015.10.003. [DOI] [PubMed] [Google Scholar]
- Ellsworth M. Ostrosky-Zeichner L. J. Fungi. 2020;6:324. doi: 10.3390/jof6040324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedmousavi S. Verweij P. E. Mouton J. W. Expert Rev. Anti-infect. Ther. 2015;13:9–27. doi: 10.1586/14787210.2015.990382. [DOI] [PubMed] [Google Scholar]
- Pena C. Suarez C. Ocampo-Sosa A. Murillas J. Almirante B. Pomar V. Aguilar M. Granados A. Calbo E. Rodriguez-Bano J. Rodriguez F. Tubau F. Oliver A. Martinez-Martinez L. Spanish Network Res Infect D. Clin. Infect. Dis. 2013;57:208–216. doi: 10.1093/cid/cit223. [DOI] [PubMed] [Google Scholar]
- Schmitt-Hoffmann A. Roos B. Heep M. Int. J. Antimicrob. Agents. 2007;29:S262. doi: 10.1016/S0924-8579(07)70828-2. [DOI] [Google Scholar]
- Hata K. Parasitol. Int. 2021;81:102278. doi: 10.1016/j.parint.2020.102278. [DOI] [PubMed] [Google Scholar]
- Denning D. W. Hope W. W. Trends Microbiol. 2010;18:195–204. doi: 10.1016/j.tim.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Xie F. Ni T. Zhao J. Pang L. Li R. Cai Z. Ding Z. Wang T. Yu S. Jin Y. Zhang D. Jiang Y. Bioorg. Med. Chem. Lett. 2017;27:2171–2173. doi: 10.1016/j.bmcl.2017.03.062. [DOI] [PubMed] [Google Scholar]
- Niwano Y. Kuzuhara N. Kodama H. Yoshida M. Miyazaki T. Yamaguchi H. Antimicrob. Agents Chemother. 1998;42:967–970. doi: 10.1128/AAC.42.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A. K. Foley K. A. Versteeg S. G. Mycopathologia. 2017;182:127–141. doi: 10.1007/s11046-016-0045-0. [DOI] [PubMed] [Google Scholar]
- Hassan N. Singh M. Sulaiman S. Jain P. Sharma K. Nandy S. Dudeja M. Ali A. Iqbal Z. ACS Omega. 2019;4:9583–9592. doi: 10.1021/acsomega.9b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley K. Gupta A. K. Versteeg S. Mays R. Villanueva E. John D. Cochrane Database Syst. Rev. 2020;1:CD012093. doi: 10.1002/14651858.CD012093.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher R. K. Nakamura N. Tavakkol A. Mycoses. 2014;57:389–393. doi: 10.1111/myc.12168. [DOI] [PubMed] [Google Scholar]
- Schafer J. J. Short W. R. Antiviral Ther. 2012;17:1495–1502. doi: 10.3851/IMP2254. [DOI] [PubMed] [Google Scholar]
- Smith S. J. Pauly G. T. Hewlett K. Schneider J. P. Hughes S. H. Chem. Biol. Drug Des. 2021;97:4–17. doi: 10.1111/cbdd.13766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmann N. A. Lang D. Saleh S. Van der Mey D. Gerisch M. Expert Opin. Drug Metab. Toxicol. 2019;15:975–984. doi: 10.1080/17425255.2019.1681968. [DOI] [PubMed] [Google Scholar]
- Das K. Clark A. D. Lewi P. J. Heeres J. de Jonge M. R. Koymans L. M. H. Vinkers H. M. Daeyaert F. Ludovici D. W. Kukla M. J. De Corte B. Kavash R. W. Ho C. Y. Ye H. Lichtenstein M. A. Andries K. Pauwels R. de Bethune M. P. Boyer P. L. Clark P. Hughes S. H. Janssen P. A. J. Arnold E. J. Med. Chem. 2004;47:2550–2560. doi: 10.1021/jm030558s. [DOI] [PubMed] [Google Scholar]
- Kang D. Sun Y. Murugan N. A. Feng D. Wei F. Li J. Jiang X. De Clercq E. Pannecouque C. Zhan P. Liu X. ACS Infect. Dis. 2020;6:2225–2234. doi: 10.1021/acsinfecdis.0c00327. [DOI] [PubMed] [Google Scholar]
- Kang D. Feng D. Ginex T. Zou J. Wei F. Zhao T. Huang B. Sun Y. Desta S. De Clercq E. Pannecouque C. Zhan P. Liu X. Acta Pharm. Sin. B. 2020;10:878–894. doi: 10.1016/j.apsb.2019.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B. Chen W. Zhao T. Li Z. Jiang X. Ginex T. Vilchez D. Luque F. J. Kang D. Gao P. Zhang J. Tian Y. Daelemans D. De Clercq E. Pannecouque C. Zhan P. Liu X. J. Med. Chem. 2019;62:2083–2098. doi: 10.1021/acs.jmedchem.8b01729. [DOI] [PubMed] [Google Scholar]
- Kang D. Wang Z. Chen M. Feng D. Wu G. Zhou Z. Jing L. Zuo X. Jiang X. Daelemans D. De Clercq E. Pannecouque C. Zhan P. Liu X. Chem. Biol. Drug Des. 2019;93:430–437. doi: 10.1111/cbdd.13429. [DOI] [PubMed] [Google Scholar]
- Lee W.-G. Chan A. H. Spasov K. A. Anderson K. S. Jorgensen W. L. ACS Med. Chem. Lett. 2016;7:1156–1160. doi: 10.1021/acsmedchemlett.6b00390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. J. Pauly G. T. Akram A. Melody K. Ambrose Z. Schneider J. P. Hughes S. H. J. Acquired Immune Defic. Syndr. 2016;72:485–491. doi: 10.1097/QAI.0000000000001031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agostini M. L. Andres E. L. Sims A. C. Graham R. L. Sheahan T. P. Lu X. Smith E. C. Case J. B. Feng J. Y. Jordan R. Ray A. S. Cihlar T. Siegel D. Mackman R. L. Clarke M. O. Baric R. S. Denison M. R. MBio. 2018;9:e00221-18. doi: 10.1128/mBio.00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel D. Hui H. C. Doerffler E. Clarke M. O. Chun K. Zhang L. Neville S. Carra E. Lew W. Ross B. Wang Q. Wolfe L. Jordan R. Soloveva V. Knox J. Perry J. Perron M. Stray K. M. Barauskas O. Feng J. Y. Xu Y. Lee G. Rheingold A. L. Ray A. S. Bannister R. Strickley R. Swaminathan S. Lee W. A. Bavari S. Cihlar T. Lo M. K. Warren T. K. Mackman R. L. J. Med. Chem. 2017;60:1648–1661. doi: 10.1021/acs.jmedchem.6b01594. [DOI] [PubMed] [Google Scholar]
- Shannon A. Nhung Thi-Tuyet L. Selisko B. Eydoux C. Alvarez K. Guillemot J.-C. Decroly E. Peersen O. Ferron F. Canard B. Antiviral Res. 2020;178:104793. doi: 10.1016/j.antiviral.2020.104793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeoye A. O. Oso B. J. Olaoye I. F. Tijjani H. Adebayo A. I. J. Biomol. Struct. Dyn. 2021;39:3469–3479. doi: 10.1080/07391102.2020.1765876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman J. A. Green E. M. Gouaux E. Nature. 2016;532:334–339. doi: 10.1038/nature17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durgam S. Chen C. Migliore R. Prakash C. Edwards J. Findling R. L. Paediatr. Drugs. 2018;20:353–363. doi: 10.1007/s40272-018-0290-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.-M. Han C. Lee S.-J. Patkar A. A. Masand P. S. Pae C.-U. Psychiatry Invest. 2015;12:155–163. doi: 10.4306/pi.2015.12.2.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich T. Bottcher H. Schiemann K. Holzemann G. Schwarz M. Bartoszyk G. D. van Amsterdam C. Greiner H. E. Seyfried C. A. Bioorg. Med. Chem. 2004;12:4843–4852. doi: 10.1016/j.bmc.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Zhu J. Hou W. Xu Y. Ji F. Wang G. Chen C. Lin C. Lin X. Li J. Zhuo C. Shao M. Psychiatry Res. 2019;281:112598. doi: 10.1016/j.psychres.2019.112598. [DOI] [PubMed] [Google Scholar]
- Alvarez-Guerra M. d'Alche-Biree F. Wolf W. A. Vargas F. Dib M. Garay R. P. Psychopharmacology. 2000;147:412–417. doi: 10.1007/s002130050010. [DOI] [PubMed] [Google Scholar]
- Radat F. Garay R. d'Alche-Biree F. Dib M. Eur. Psychiatry. 2000;15:380S. doi: 10.1016/S0924-9338(00)94626-1. [DOI] [Google Scholar]
- Tervo A. J. Nyronen T. H. Ronkko T. Poso A. J. Comput.-Aided Mol. Des. 2003;17:797–810. doi: 10.1023/B:JCAM.0000021831.47952.a7. [DOI] [PubMed] [Google Scholar]
- Di Bonaventura C. Labate A. Maschio M. Meletti S. Russo E. Expert Opin. Pharmacother. 2017;18:1751–1764. doi: 10.1080/14656566.2017.1392509. [DOI] [PubMed] [Google Scholar]
- Wheless J. Wechsler R. T. Lancman M. Aboumatar S. Patten A. Malhotra M. Epilepsia Open. 2021;6:79–89. doi: 10.1002/epi4.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelshanskaya M. V. Singh A. K. Sampson J. M. Narangoda C. Kurnikova M. Sobolevsky A. I. Neuron. 2016;91:1305–1315. doi: 10.1016/j.neuron.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibi S. Ueno K. Nagato S. Kawano K. Ito K. Norimine Y. Takenaka O. Hanada T. Yonaga M. J. Med. Chem. 2012;55:10584–10600. doi: 10.1021/jm301268u. [DOI] [PubMed] [Google Scholar]
- Barlesi F. Mazieres J. Merlio J.-P. Debieuvre D. Mosser J. Lena H. Ouafik L. H. Besse B. Rouquette I. Westeel V. Èscande F. Monnet I. Lemoine A. Veillon R. Blons H. Audigier-Valette C. Bringuier P.-P. Lamy R. Beau-Faller M. Pujol J.-L. Sabourin J.-C. Penault-Llorca F. Denis M. G. Lantuejoul S. Morin F. Quan T. Missy P. Langlais A. Milleron B. Cadranel J. Soria J.-C. Zalcman G. Biomarkers France C. Lancet. 2016;387:1415–1426. doi: 10.1016/S0140-6736(16)00004-0. [DOI] [PubMed] [Google Scholar]
- Soda M. Choi Y. L. Enomoto M. Takada S. Yamashita Y. Ishikawa S. Fujiwara S.-i. Watanabe H. Kurashina K. Hatanaka H. Bando M. Ohno S. Ishikawa Y. Aburatani H. Niki T. Sohara Y. Sugiyama Y. Mano H. Nature. 2007;448:U561–U563. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- Choi Y. L. Takeuchi K. Soda M. Inamura K. Togashi Y. Hatano S. Enomoto M. Hamada T. Haruta H. Watanabe H. Kurashina K. Hatanaka H. Ueno T. Takada S. Yamashita Y. Sugiyama Y. Ishikawa Y. Mano H. Cancer Res. 2008;68:4971–4976. doi: 10.1158/0008-5472.CAN-07-6158. [DOI] [PubMed] [Google Scholar]
- Sakamoto H. Tsukaguchi T. Hiroshima S. Kodama T. Kobayashi T. Fukami T. A. Oikawa N. Tsukuda T. Ishii N. Aoki Y. Cancer Cell. 2011;19:679–690. doi: 10.1016/j.ccr.2011.04.004. [DOI] [PubMed] [Google Scholar]
- He M. Li W. Zheng Q. Zhang H. J. Cell. Biochem. 2018;119:5332–5342. doi: 10.1002/jcb.26666. [DOI] [PubMed] [Google Scholar]
- Katayama R. Shaw A. T. Khan T. M. Mino-Kenudson M. Solomon B. J. Halmos B. Jessop N. A. Wain J. C. Yeo A. T. Benes C. Drew L. Saeh J. C. Crosby K. Sequist L. V. Iafrate A. J. Engelman J. A. Sci. Transl. Med. 2012;4:120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama R. Sakashita T. Yanagitani N. Ninomiya H. Horiike A. Friboulet L. Gainor J. F. Motoi N. Dobashi A. Sakata S. Tambo Y. Kitazono S. Sato S. Koike S. Iafrate A. J. Mino-Kenudson M. Ishikawa Y. Shaw A. T. Engelman J. A. Takeuchi K. Nishio M. Fujita N. EBioMedicine. 2016;3:54–66. doi: 10.1016/j.ebiom.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. W. Richardson P. F. Bailey S. Brooun A. Burke B. J. Collins M. R. Cui J. J. Deal J. G. Deng Y.-L. Dinh D. Engstrom L. D. He M. Hoffman J. Hoffman R. L. Huang Q. Kania R. S. Kath J. C. Lam H. Lam J. L. Le P. T. Lingardo L. Liu W. McTigue M. Palmer C. L. Sach N. W. Smeal T. Smith G. L. Stewart A. E. Timofeevski S. Zhu H. Zhu J. Zou H. Y. Edwards M. P. J. Med. Chem. 2014;57:4720–4744. doi: 10.1021/jm500261q. [DOI] [PubMed] [Google Scholar]
- Lin J. J. Liu S. V. McCoach C. E. Zhu V. W. Tan A. C. Yoda S. Peterson J. Do A. Prutisto-Chang K. Dagogo-Jack I. Sequist L. V. Wirth L. J. Lennerz J. K. Hata A. N. Mino-Kenudson M. Nardi V. Ou S. H. I. Tan D. S. W. Gainor J. F. Ann. Oncol. 2020;31:1725–1733. doi: 10.1016/j.annonc.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford D. Larkins E. Mushti S. L. Rodriguez L. Skinner A. M. Helms W. S. Price L. S. L. Zirkelbach J. F. Li Y. Liu J. Charlab R. Turcu F. R. Liang D. Ghosh S. Roscoe D. Philip R. Zack-Taylor A. Tang S. Kluetz P. G. Beaver J. A. Pazdur R. Theoret M. R. Singh H. Clin. Cancer Res. 2021;27:2130–2135. doi: 10.1158/1078-0432.CCR-20-3558. [DOI] [PubMed] [Google Scholar]
- Roskoski, Jr. R. Pharmacol. Res. 2021:105463. doi: 10.1016/j.phrs.2021.105463. doi: 10.1016/j.phrs.2021.105463. [DOI] [PubMed] [Google Scholar]
- Subbiah V. Shen T. Terzyan S. S. Liu X. Hu X. Patel K. P. Hu M. Cabanillas M. Behrang A. Meric-Bernstam F. Vo P. T. T. Mooers B. H. M. Wu J. Ann. Oncol. 2021;32:261–268. doi: 10.1016/j.annonc.2020.10.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V. Gohil N. Ramirez-Garcia R. J. Cell. Biochem. 2018;119:2003–2011. doi: 10.1002/jcb.26361. [DOI] [PubMed] [Google Scholar]
- Jafarzadeh A. Nemati M. Khorramdelazad H. Hassan Z. M. Int. Immunopharmacol. 2019;70:156–166. doi: 10.1016/j.intimp.2019.02.026. [DOI] [PubMed] [Google Scholar]
- Rafiee S. Asadollahi K. Riazi G. Ahmadian S. Saboury A. A. ACS Chem. Neurosci. 2017;8:2676–2682. doi: 10.1021/acschemneuro.7b00230. [DOI] [PubMed] [Google Scholar]
- O'Leary F. Samman S. Nutrients. 2010;2:299–316. doi: 10.3390/nu2030299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S. Mahalle N. Greibe E. Ostenfeld M. S. Heegaard C. W. Nexo E. Fedosov S. N. Nutrients. 2019;11:2382. doi: 10.3390/nu11102382. [DOI] [PMC free article] [PubMed] [Google Scholar]