

Abstract
Background. There is increasing interest in methods to more rapidly and cost-efficiently investigate drugs that are approved for clinical use in the treatment of another condition. Exenatide is a type 2 diabetes treatment that has been shown to have neuroprotective/neurorestorative properties in preclinical models of neurodegeneration.
Methods. As a proof of concept, using a single-blind trial design, we evaluated the progress of 45 patients with moderate Parkinson’s disease (PD), randomly assigned to receive subcutaneous exenatide injection for 12 months or to act as controls. Their PD was compared after overnight withdrawal of conventional PD medication using blinded video assessment of the Movement Disorders Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS), together with several nonmotor tests, at baseline, 6 months, and 12 months and after a further 2-month washout period (14 months).
Results. Exenatide was well tolerated, although weight loss was common and l-dopa dose failures occurred in a single patient. Single-blinded rating of the exenatide group suggested clinically relevant improvements in PD across motor and cognitive measures compared with the control group. Exenatide-treated patients had a mean improvement at 12 months on the MDS-UPDRS of 2.7 points, compared with mean decline of 2.2 points in control patients (P = 0.037).
Conclusion. These results demonstrate a potential cost-efficient approach through which preliminary clinical data of possible biological effects are obtainable, prior to undertaking the major investment required for double-blind trials of a potential disease-modifying drug in PD.
Trial registration. Clinicaltrials.gov NCT01174810.
Funding. Cure Parkinson’s Trust.
Introduction
The management of Parkinson’s disease (PD) consists of therapies that aim to relieve the symptoms of the condition at its various stages. There are no approaches universally accepted to modify the progressive course of the disease, despite major financial investment into double-blind randomized trials of agents showing neuroprotective effects in laboratory models (1). As a result, there is increased caution, from both the commercial sector and governmental and charitable funding agencies, when making decisions regarding further large-scale investments into even the most promising novel drug therapies.
There has nevertheless been recent interest in the use of agonists for the glucagon-like peptide–1 (GLP-1) receptor as therapeutic agents in neurodegenerative diseases (2). The first of these, exendin-4, was discovered in the saliva of the Gila monster (3) and found to have beneficial effects on glucose homeostasis through stimulation of glucose level–dependent insulin release, β islet cell proliferation, reduction of β islet cell apoptosis, and weight loss (4, 5). The synthetic version of exendin-4, exenatide, was granted a license for the treatment of type 2 diabetes mellitus (T2DM) in 2005.
GLP-1 receptors have also been identified throughout the brain, and in vitro work suggesting neurotrophic and neuroprotective effects of exenatide (6, 7) has been extended to in vivo rodent models showing beneficial effects on learning and memory (8), as well as neuroprotective and neurorestorative effects after administration of a wide range of toxins to the dopaminergic system (9–12). However, the mechanism of any neuroprotective action of exenatide is uncertain, with suggestions that it may be neurotrophic (7), may act as an antiinflammatory agent (12), may facilitate neurogenesis (10), or may stimulate mitochondrial biogenesis (13). Interest in the possible therapeutic effects of GLP-1 agonists extends across medical disciplines, and several trials are currently in progress, including an evaluation of its effects as a neuroprotective agent in Alzheimer’s disease ( www.clinicaltrials.gov).
The current trial design was conceived following feedback from both commercial and charitable organizations, which confirmed the impression that the risks associated with investment into potential neuroprotective agents need to be mitigated via the preliminary collection of cost-efficient (open-label) data in the first instance. In this context, the current study was designed as a proof of principle (14); i.e., with the aims of collecting rapid and cost-efficient data regarding the tolerability of exenatide in patients with PD and providing preliminary indications whether the major neuroprotective and neurorestorative effects of exenatide seen in the animal models might be replicable in human individuals with PD.
Results
Patients
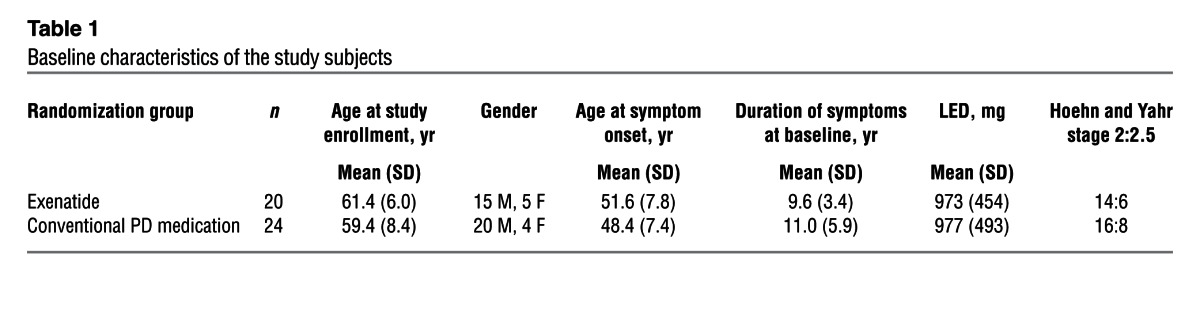
Of the 45 patients recruited, 1 patient randomized to exenatide dropped out before first follow-up, and was therefore replaced per the study protocol and not included in the final analysis (Figure 1). The baseline demographic data for the 44 participants included in the final analysis are presented in Table 1. Clinical data were missing for 2 individuals (1 individual per group), both for their 14-month visits. The first 10 patients allocated to exenatide treatment successfully had [123I]FP-CIT SPECT scans (DaTscan) at baseline and at 12 months. Based on the remaining doses of exenatide remaining in each exenatide pen device at each visit, compliance was judged to be very high for all participants.
Figure 1. Random assignments to treatment, completion of the trial, and reasons for not completing it.

Patients withdrawing from the trial prior to the 3-month visit were replaced, and new recruits were randomly allocated to the 2 groups.
Table 1.
Baseline characteristics of the study subjects
Clinical outcomes
Blinded video rating of MDS-UPDRS part 3 in the practically defined “off-medication” condition.
Patients allocated to the exenatide group had a mean improvement at 12 months of 2.7 points (SD, 7.7) on the Movement Disorders Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) part 3, while controls had a mean decline of 2.2 points (SD, 7.3), for a difference of 4.9 points (95% CI, 0.3–9.4; P = 0.037; Table 2). At 14 months, the exenatide group had a mean improvement of 1.7 points (SD, 7.4), while controls had a mean decline of 2.8 points (SD, 6.7), for a difference of 4.4 points (95% CI, 0.2–8.7; P = 0.042). These scores did not include any changes detected in limb or neck rigidity, which cannot be reliably rated on video. Addition of open-label rating of rigidity scores to the blinded data equated to an improvement of 1.8 points (SD, 8.7) in the exenatide group at 12 months, compared with a decline of 5.3 points (SD, 8.3) in the control group (difference, 7.0 points; 95% CI, 1.8–12.2; P = 0.009), and an improvement of 0.8 points (SD, 8.7) in the exenatide group at 14 months, compared with a decline of 6.4 points (SD, 7.8) in the control group (difference, 7.2 points; 95% CI, 2.1–12.2; P = 0.006; Figure 2).
Table 2.
Changes in MDS-UPDRS score between baseline and month 14
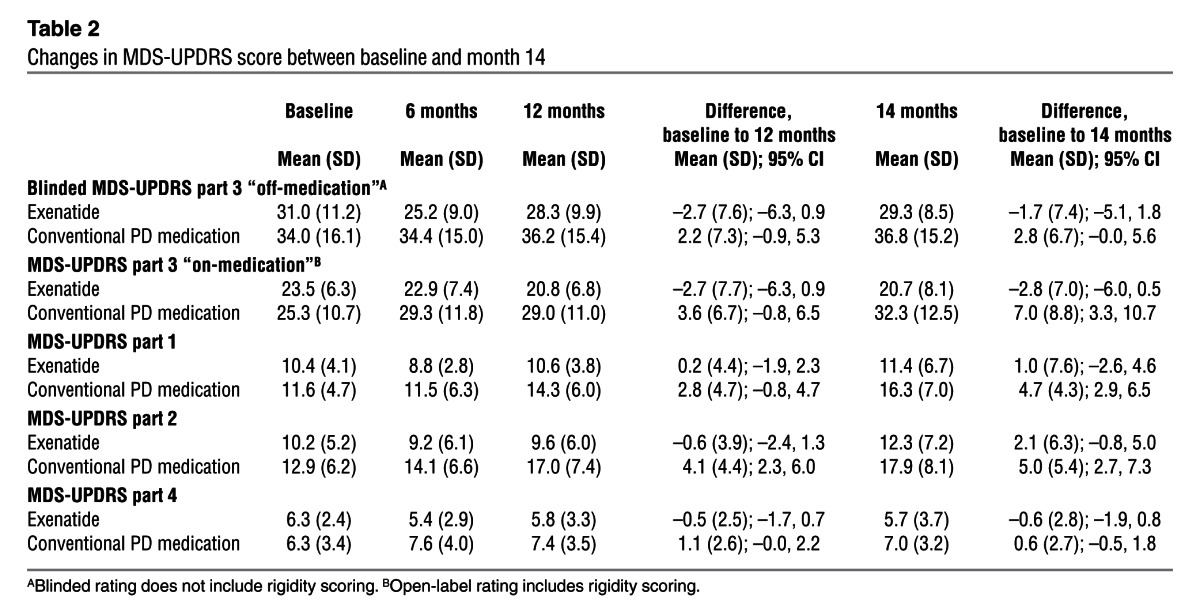
Figure 2. Change from baseline in the MDS-UPDRS part 3 score by study visit.

Data represent mean ± SEM.
Secondary outcome measures.
Open-label rating of MDS-UPDRS part 3 “on-medication” showed a difference favoring patients treated with exenatide compared with the control group. The mean improvement in “on-medication” scores in the exenatide group (2.7 points) was unchanged between the 12- and 14-month visits. In contrast, individuals in the control group deteriorated over the total 14-month trial period by 7 points. There was a significant difference or nonsignificant trend favoring exenatide in MDS-UPDRS parts 1, 2, and 4 at both the 12- and 14-month time points (Table 2).
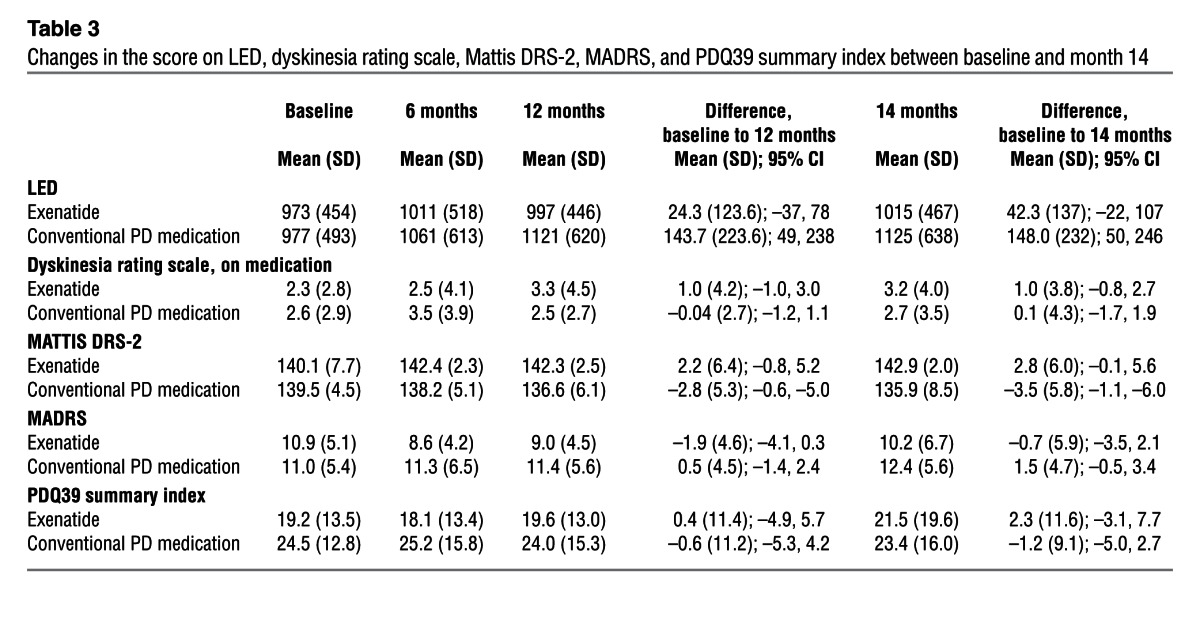
At both 12 and 14 months, a significant advantage in the Mattis dementia rating scale–2 (Mattis DRS-2) was seen in patients treated with exenatide, with a mean improvement of 2.8 points at 14 months compared with deterioration by a mean of 3.5 points in control patients (difference, 6.3 points; 95% CI, 2.7–9.9; P = 0.001; Figure 3 and Table 3). There were also general trends for improvement in the timed tests and l-dopa equivalent dose (LED), but no significant difference in Montogomery-Asberg depression rating scale (MADRS) scores or Parkinson Disease Questionnaire–39 (PDQ39) summary index scores between the 2 groups (Table 3).
Figure 3. Change from baseline in the Mattis DRS-2 score by study visit.

Data represent mean ± SEM.
Table 3.
Changes in the score on LED, dyskinesia rating scale, Mattis DRS-2, MADRS, and PDQ39 summary index between baseline and month 14
Safety and tolerability.
4 patients withdrew/dropped out from the study, 3 from the group randomized to exenatide and 1 from the conventional PD medication group. Of the 3 patients in the exenatide group, 1 withdrew due to worsening PD (recurrent l-dopa dose failures) prior to the first follow-up visit. Exenatide is known to cause slowing of gastric emptying and is the most likely explanation for this observation in this patient. 2 further patients withdrew from treatment: the first at 9 months due to dysgeusia combined with subjective PD deterioration, and the second at 10 months due to excessive weight loss (despite reduction to the 5-μg dose). 1 patient randomized to the conventional PD medication group withdrew from the study at 12 months due to deteriorating PD and incapacity to attend the trial clinic in the “off-medication” state. All serious adverse events, together with adverse events occurring in more than 1 subject, are listed in Table 4.
Table 4.
Adverse events reported by trial participants

No clinically relevant changes in ECG, hematological, or biochemical indices were observed. Weight loss and nausea were more common in the patient group treated with exenatide. Patients on exenatide lost a mean of 3.2 kg (SD, 3.9; range, 3.5-kg increase to 12.3-kg decrease) over 12 months, necessitating 2 patients to reduce to the 5-μg dose. Control group patients lost a mean of 0.8 kg (SD, 3.3; range, 5.5-kg increase to 7.8-kg decrease) over 12 months. There was a greater increase in the mean dyskinesia rating scale score in exenatide versus control patients at both 12 and 14 months. This necessitated lowering of LED in 5 exenatide patients, whereas 8 exenatide patients had unchanged dopaminergic medication and 7 patients had increased dopaminergic medication over the period of study.
Substudy
All exenatide group patients had profoundly abnormal [123I]FP-CIT SPECT scans at baseline, with some variation in severity of presynaptic dopaminergic deficit. 2 patients with severe baseline presynaptic deficits had minor improvement in [123I]FP-CIT uptake in all basal ganglia subregions at 12 months. 1 individual had deterioration in all subregions. Mean values for absolute and percent changes in [123I]FP-CIT activity showed minimal change in all basal ganglia subregions in the exenatide group at 12 months.
Discussion
This is the first trial to report tolerability and pilot data of the biological effects of exenatide in patients with a neurodegenerative disease. In view of the single-blind design, we cannot exclude placebo effects being responsible for the observed differences between patients treated with exenatide and controls, and these data should not be interpreted as evidence of symptomatic efficacy or neuroprotection. Given the complex design of the exenatide pen device, the cost of manufacture of a matched placebo, and the absence of commercial sponsorship, this weakness was unavoidable, and these data should be considered as proof of principle/proof of concept only. Nevertheless, the trial design allowed the collection of data in a very cost-efficient manner, and the results demonstrated that exenatide was generally well tolerated by PD patients and that clinically relevant differences emerged between the 2 groups that persisted beyond the 12-month exposure to the study drug, which may potentially reflect biological activity.
In an attempt to improve the cost-efficiency of trials of potential neuroprotective agents, previous investigators have used “futility” designs in order to compare disease progression in individuals assigned to experimental agents against the expected natural history of the disease, based on either historical or contemporary control data (15, 16). These studies have highlighted the difficulties in selecting agents for major investment, given the presence of highly variable rates of placebo responses that can occur. The variable nature of the placebo response is a particularly important issue for complex or invasive interventions in which placebo versions of the licensed product are not readily available/represent additional significant expense, which can thus hinder the conduct and interpretation of even small phase 2 double-blind trials. In the current trial, we adopted a proof of principle approach to provide preliminary data regarding the tolerability of exenatide in a small number of patients with PD, as well as to collect pilot data with respect to possible biological effects, in order to help justify the larger investment required to initiate a larger double-blind, placebo-controlled study and assist in sample size calculations. We chose not to set any futility threshold a priori, but instead chose to use a contemporary group of PD controls and to continue follow-up for a sufficiently long period to allow inevitable placebo effects to at least begin to diminish.
There are multiple issues in the optimal design of trials that aim to identify agents with neuroprotective effects that have been previously highlighted, including the prioritization of drugs for study, the optimal trial design and duration, the ideal outcome measures, and the optimal group of patients for study (1). In the current study, the inclusion and exclusion criteria for patient selection were chosen on a pragmatic basis to minimize the risk of including patients with non-PD tremor or atypical Parkinsonism, with the understanding that patients with advanced disease would be likely to have fewer salvageable dopaminergic neurons. We also wished to determine whether this drug was tolerable in moderate-stage PD patients on l-dopa, given that it is unlikely that any neuroprotective agent will ever be able to entirely replace the need for symptomatic therapy. Since exenatide is also being evaluated as an agent to influence cognitive decline, our inclusion criteria allowed us to evaluate the possible influence of exenatide on cognition. Given that exenatide has not previously been given to patients with PD, the design of this trial included a washout period to also allow for preliminary distinctions between possible symptomatic effects and possible disease-modifying effects. However, the inclusion of a 2-month washout period provided information on short-term symptomatic effects only. Long-duration symptomatic effects may persist beyond this period and cannot be excluded using the current study design. This is particularly relevant given the increase in l-dopa–induced dyskinesia (LID) seen in the exenatide-treated group, which necessitated reduction in l-dopa doses in 5 patients. The possibility of an interaction between exenatide and conventional dopaminergic replacement must be considered in future study designs.
A further caveat must be made in the interpretation of these data given the small sample size. Despite randomization, minor differences in the baseline characteristics of the treated and control groups can influence subsequent disease progression. In a larger sample, randomization would be more likely to balance the treated and untreated groups. In the current trial, we attempted to minimize this chance variation by stratified randomization according to baseline disease severity. While there were no significant differences between the 2 groups at baseline, the control group had slightly longer disease duration than that of exenatide-treated patients.
Weight loss is an important concern and prevented trial completion in 1 individual. This was fully reversible on cessation of the drug. Gastrointestinal symptoms are a common side effect of exenatide and also in the PD population, but did not compromise trial participation in any individual. Patients generally tolerated the pen injection device well, and none of the serious adverse events observed were considered to be reactions to exenatide. The frequency of the adverse events in the exenatide group was similar to that seen in the previous clinical trials of exenatide in diabetes patients (4, 5).
Bearing in mind these limitations, our data provided preliminary information about exenatide tolerability in PD and allowed consideration of the size of the biological effects seen in comparison to previous trials of patients on placebo medications. The absolute size of the difference in PD severity between the exenatide-treated and untreated groups using blinded rating was modest (4.9 points in MDS-UPDRS part 3), although this value excluded the additional effects on rigidity scores, which were only evaluated using open-label rating. Inclusion of the rigidity scoring made by the unblinded investigator equated to a 7.0-point difference in MDS-UPDRS score at 12 months and a 7.2-point difference at 14 months. Adding the blinded video rating of MDS-UPDRS part 3 to the differences seen in parts 1, 2, and 4 of the scale equated to a 13.8-point advantage in favor of exenatide at 12 months and a 12.3-point advantage at 14 months. The changes detected in MDS-UPDRS scores were also largely reflected in the timed motor tests. Aside from the changes in MDS-UPDRS scores, there was also divergence in cognitive performance between the groups, with a 5-point advantage in the Mattis DRS-2 at 12 months that persisted as a 6.3-point advantage at 14 months. However, there were no significant changes detected in depression or subjective ratings of quality of life (i.e., PDQ39 summary index).
There was no change in [123I]FP-CIT SPECT scan appearances over a 12-month period. Given the quite advanced stage of disease suggested by the baseline levels of [123I]FP-CIT uptake, this may simply reflect a slower rate of decline of PD than occurs in the first few years. The small mean improvements in [123I]FP-CIT uptake in 2 patients is out of keeping with the natural history of PD. These patients both experienced LID during the course of the trial that responded to lowering of LED.
The data presented herein demonstrated that clinically informative data can be obtained in a very cost-efficient manner as part of the process of selecting drugs for future study as potential neuroprotective agents in PD. These data support further, double-blind trials of exenatide as a potential disease-modifying drug in PD, which will incur substantially greater costs than were required for the current study. It is arguable that prevention of deterioration is more achievable in the earlier stages of PD, when a greater number of dopaminergic neurons are still viable. While the major cohort of interest for future trials may be subjects with early PD, the data presented here indicate that further investigation of exenatide as a treatment in later stages of PD may also be warranted. The changes on the Mattis DRS-2 scale suggest that trials of the drug in the degenerative dementias would also be of interest.
Methods
Study design.
This study was designed to collect preliminary safety and efficacy data regarding the long-term biological effects of exenatide in patients with treated PD. This was an investigator-led, charity-funded project without commercial sponsorship. The licensed product of exenatide is manufactured as a pen device designed for self administration of 60 doses (i.e., sufficient for 1 month’s treatment). In view of the prohibitive costs associated with manufacture of (QP released) placebo versions of the exenatide pens, these were not available for the purposes of this trial, which was necessarily configured to be open-label from the patient’s perspective. Given the progressive nature of PD, a parallel-group, randomized, controlled design was chosen, with follow-up over a 12-month period. In the absence of any previous evidence confirming or excluding possible short-term symptomatic benefits of exenatide in PD, a 2-month washout period was included beyond the end of study drug exposure. The overall aim was to collect data from a range of outcome measures that, if positive, would provide support/reassurance to underpin the investment needed to perform double-blind, placebo-controlled trials of exenatide in PD. See clinical protocol in the supplemental material (supplemental material available online with this article; doi: 10.1172/JCI68295DS1).
Patients.
The patient group was chosen to represent typical, l-dopa–responsive PD, in whom the severity of underlying PD could be assessed during an early-morning “off-medication” examination. Patients were eligible for enrollment if they had PD meeting Queen Square Brain bank criteria at Hoehn and Yahr stage 2 to 2.5 in the “on-medication” state; they had at least 5 years duration of symptoms; they were between 45 and 70 years of age; they were on l-dopa treatment with wearing-off phenomenon; they were on stable PD medication regimes; they had “off-medication” MDS-UPDRS part 3 scores greater than 15 points; and they had at least 33% improvement in response to l-dopa. Female participants had to be free of child-bearing potential and were not pregnant or breast feeding. Male participants had to use adequate contraception (given the absence of long-term safety data regarding exenatide with respect to conception).
Participants were excluded if they had another suspected cause for their parkinsonism; a known abnormality on structural brain imaging that might compromise trial participation; concurrent dementia (judged clinically using a cutoff score lower than 120 on the Mattis DRS-2); severe depression (defined by a score greater than 16 on the MADRS); neuroleptic exposure in the preceding 6 months; diabetes mellitus; previous neurosurgery; a history of severe renal disease, cardiac disease, gastrointestinal disease, alcoholism, or pancreatitis.
Randomization.
Subjects were randomly assigned to 2 groups (Figure 1): (a) exenatide added to conventional PD treatment group and (b) conventional PD treatment only (control) group. Randomization was stratified by Hoehn and Yahr stage (2, low; 2.5, high) to help ensure similar baseline disability between the 2 groups. Recruitment ended when 20 subjects randomized to each group had reached their 3-month follow-up stage. As a result, 21 patients were randomized to the exenatide group and 24 patients were randomized to conventional PD treatment only.
Subjects in the exenatide group were supplied with a 5-μg exenatide pen device (Byetta 5 μg) and taught how to self-administer twice-daily injections for 1 month, then supplied with 10-μg exenatide pen device (Byetta 10 μg) to self-administer twice-daily injections for the subsequent 11 months. This schedule has been associated with lower rates of nausea than immediate introduction of the 10-μg dose (17). Following subcutaneous administration, exenatide reaches median peak plasma concentrations in 2 hours. Similar exposure is achieved with subcutaneous administration of exenatide in the abdomen, thigh, or arm (18). Exenatide has the ability to cross the blood-brain barrier after peripheral administration (19).
Bioavailability of the 10-μg dose compares favorably to the efficacious plasma levels in the rodent models (200 pg/ml) (20). Patients in both groups continued any licensed PD medication after enrollment and throughout the trial according to the judgement of their treating neurologists. No adjustment of PD medications was made at trial visits unless clinically urgent.
Clinical evaluation.
All patients had evaluations at baseline, 6 months, 12 months, and 14 months (i.e., allowing a 2-month washout phase of exenatide). Assessments of PD severity using MDS-UPDRS part 3 were made “off-medication” after an overnight period and were video recorded to allow objective rating of PD disability by observers blinded to randomization outcomes. Each patient video was rated by the same blinded clinician at each time point. All clinicians were experienced at evaluating PD patients and had successfully completed the official training module of the MDS-UPDRS. Sustained-release dopamine agonist medications were withdrawn for 24 hours, while the withdrawal period for l-dopa was 12 hours.
All subscores of the MDS-UPDRS were completed, with the part 3 motor subscore repeated during the “on-medication” state. Rush dyskinesia rating scale and timed motor tests were performed. Calculation of LED was performed using published formulae (21).
While “on-medication,” each subject was assessed with the Mattis DRS-2, the MADRS, the PDQ39, the Non-Motor Symptoms (NMS) Questionnaire, the EuroQoL-5D (EQ-5D), and the SCOPA SLEEP and SCOPA Autonomic (SCOPA AUT) questionnaires. The Smell Identification test (Sensonics Inc.) was performed at baseline and 12-month visits only. Safety assessments included measurements of vital signs and weight, electrocardiography, laboratory blood tests, and recording of adverse events. All patients were also contacted at 1, 3, and 9 months to ensure comprehensive reporting of adverse events. Patients were asked to return used pens at each visit, and the remaining doses were counted as a measure of compliance.
Substudy.
The first 10 patients randomized to receive exenatide had an [123I]FP-CIT SPECT scan performed at baseline and repeated at 12 months, with the aim of identifying whether this may serve as a biomarker of responsiveness to exenatide. [123I]FP-CIT uptake was quantified as a ratio of specific to nonspecific binding in 3 regions of interest in each hemisphere — the caudate, putamen and striatum as a whole — using validated quantitative software (22). Absolute values of uptake ratio in each region of interest were recorded at each time point, enabling the change in absolute activity as well as the percent change in activity over the 12-month interval to be calculated.
Statistics.
The data presentation and statistical tests performed in this study have been provided to help guide planning of future placebo-controlled trials and are thus restricted to the main outcomes of interest. PD patients on stable treatment regimens decline at a rate of approximately 3 UPDRS motor points per year (SD, 6.8) (23), although this can vary according to stage of disease (24). PD trials using change in “off-medication” motor UPDRS scores as an outcome measure have shown similar rates of decline (1.7 points after 6 months) among patients on “best medical treatment” (25). The planned patient sample size (n = 40) in this trial had 90% power to detect a difference of 7 UPDRS points between the exenatide and control groups at a significance level of 5%. All individuals completing a single follow-up were included in final analysis, as per protocol. “Last observation carried forward” was used for participants with missing data. Data were tested for normality using the Shapiro-Wilk test. Differences between treated and untreated groups with respect to change in scores from baseline to follow-up were analyzed using 2-sided t tests. All data were analyzed using Stata version 8.0.
Study approval.
The trial was conducted at the National Hospital for Neurology and Neurosurgery (London, United Kingdom), with trial sponsorship and monitoring by University College London. The protocol was approved by a Research Ethics committee, the Administration of Radioactive Substances Advisory Committee (ARSAC), and the UK Medicines Health Regulatory Agency, and all patients signed informed consent. A trial management group reviewed trial conduct and all safety data. The trial was registered at www.clinicaltrials.gov (trial ID NCT01174810).
Supplementary Material
Acknowledgments
We thank the PD patients and their families who participated in and supported the conduct of this trial; Gareth Ambler, who provided additional statistical advice during the writing of the trial protocol; Gurjinder Kahlon, Suzanne Hodgson, Farhat Gilani, Salwa Beydoun, and Helen Cadiou from the UCL Joint Research Office; Wendy Waddington, for providing Medical Physics support; and all clinicians referring patients to the trial. We owe a debt of gratitude to the work of Helen Matthews at CPT. The work was undertaken at UCL/UCLH, which is partly funded by the Department of Health NIHR Biomedical Research Centres funding scheme. T. Foltynie is supported by the Parkinson’s Appeal.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Role of funding source: This study was funded by the Cure Parkinson’s Trust. Corresponding author Thomas Foltynie had control of and access to all data collected and was responsible for the decision to submit for publication.
Citation for this article: J Clin Invest. 2013;123(6):2730–2736. doi:10.1172/JCI68295.
See the related Commentary beginning on page 2364.
References
- 1.Olanow CW, Kieburtz K, Schapira AH. Why have we failed to achieve neuroprotection in Parkinson’s disease? Ann Neurol. 2008;64(suppl 2):S101–S110. doi: 10.1002/ana.21461. [DOI] [PubMed] [Google Scholar]
- 2.Aviles-Olmos I, Limousin P, Lees A, Foltynie T. Parkinson’s disease, insulin resistance and novel agents of neuroprotection. Brain. 2013;136(pt 2):374–384. doi: 10.1093/brain/aws009. [DOI] [PubMed] [Google Scholar]
- 3.Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem. 1992;267(11):7402–7405. [PubMed] [Google Scholar]
- 4.Buse JB, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care. 2004;27(11):2628–2635. doi: 10.2337/diacare.27.11.2628. [DOI] [PubMed] [Google Scholar]
- 5.DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care. 2005;28(5):1092–1100. doi: 10.2337/diacare.28.5.1092. [DOI] [PubMed] [Google Scholar]
- 6.Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther. 2002;302(3):881–888. doi: 10.1124/jpet.102.037481. [DOI] [PubMed] [Google Scholar]
- 7.Perry T, et al. A novel neurotrophic property of glucagon-like peptide 1: a promoter of nerve growth factor-mediated differentiation in PC12 cells. J Pharmacol Exp Ther. 2002;300(3):958–966. doi: 10.1124/jpet.300.3.958. [DOI] [PubMed] [Google Scholar]
- 8.During MJ, et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med. 2003;9(9):1173–1179. doi: 10.1038/nm919. [DOI] [PubMed] [Google Scholar]
- 9.Harkavyi A, Abuirmeileh A, Lever R, Kingsbury AE, Biggs CS, Whitton PS. Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson’s disease. J Neuroinflammation. 2008;5:19. doi: 10.1186/1742-2094-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertilsson G, et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J Neurosci Res. 2008;86(2):326–338. doi: 10.1002/jnr.21483. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci U S A. 2009;106(4):1285–1290. doi: 10.1073/pnas.0806720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim S, Moon M, Park S. Exendin-4 protects dopaminergic neurons by inhibition of microglial activation and matrix metalloproteinase-3 expression in an animal model of Parkinson’s disease. J Endocrinol. 2009;202(3):431–439. doi: 10.1677/JOE-09-0132. [DOI] [PubMed] [Google Scholar]
- 13.Fan R, Li X, Gu X, Chan JC, Xu G. Exendin-4 protects pancreatic beta cells from human islet amyloid polypeptide-induced cell damage: potential involvement of AKT and mitochondria biogenesis. Diabetes Obes Metab. 2010;12(9):815–824. doi: 10.1111/j.1463-1326.2010.01238.x. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt B. Proof of Principle studies. Epilepsy Res. 2006;68(1):48–52. doi: 10.1016/j.eplepsyres.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 15.NINDS NET-PD Investigators. A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology. 2006;66(5):664–671. doi: 10.1212/01.wnl.0000201252.57661.e1. [DOI] [PubMed] [Google Scholar]
- 16.NINDS NET-PD Investigators. A randomized clinical trial of coenzyme Q10 and GPI-1485 in early Parkinson disease. Neurology. 2007;68(1):20–28. doi: 10.1212/01.wnl.0000250355.28474.8e. [DOI] [PubMed] [Google Scholar]
- 17.Fineman MS, Shen LZ, Taylor K, Kim DD, Baron AD. Effectiveness of progressive dose-escalation of exenatide (exendin-4) in reducing dose-limiting side effects in subjects with type 2 diabetes. Diabetes Metab Res Rev. 2004;20(5):411–417. doi: 10.1002/dmrr.499. [DOI] [PubMed] [Google Scholar]
- 18.Kolterman OG, et al. Pharmacokinetics, pharmacodynamics, and safety of exenatide in patients with type 2 diabetes mellitus. Am J Health Syst Pharm. 2005;62(2):173–181. doi: 10.1093/ajhp/62.2.173. [DOI] [PubMed] [Google Scholar]
- 19.Kastin AJ, Akerstrom V. Entry of exendin-4 into brain is rapid but may be limited at high doses. Int J Obes Relat Metab Disord. 2003;27(3):313–318. doi: 10.1038/sj.ijo.0802206. [DOI] [PubMed] [Google Scholar]
- 20.Calara F, et al. A randomized, open-label, crossover study examining the effect of injection site on bioavailability of exenatide (synthetic exendin-4). Clin Ther. 2005;27(2):210–215. doi: 10.1016/j.clinthera.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 21.Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE. Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov Disord. 2010;25(15):2649–2653. doi: 10.1002/mds.23429. [DOI] [PubMed] [Google Scholar]
- 22.Koch W, Radau PE, Hamann C, Tatsch K. Clinical testing of an optimized software solution for an automated, observer-independent evaluation of dopamine transporter SPECT studies. J Nucl Med. 2005;46(7):1109–1118. [PubMed] [Google Scholar]
- 23.Holloway RG, et al. Pramipexole vs levodopa as initial treatment for Parkinson disease: a 4-year randomized controlled trial. Arch Neurol. 2004;61(7):1044–1053. doi: 10.1001/archneur.61.7.1044. [DOI] [PubMed] [Google Scholar]
- 24.Schrag A, Dodel R, Spottke A, Bornschein B, Siebert U, Quinn NP. Rate of clinical progression in Parkinson’s disease. A prospective study. Mov Disord. 2007;22(7):938–945. doi: 10.1002/mds.21429. [DOI] [PubMed] [Google Scholar]
- 25.Weaver FM, et al. Bilateral deep brain stimulation vs best medical therapy for patients with advanced Parkinson disease: a randomized controlled trial. JAMA. 2009;301(1):63–73. doi: 10.1001/jama.2008.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.