

Abstract
Base excision repair plays an important role in preventing mutations associated with the common product of oxidative damage, 8-oxoguanine. Recent structural studies have shown that 8-oxoguanine glycosylases use an intricate series of steps to efficiently search and locate 8-oxoguanine lesions within the multitude of undamaged bases. The importance of prevention of mutations associated with 8-oxoguanine has also been illustrated by direct connections between defects in the BER glycosylase MUTYH and colorectal cancer. In addition, the properties of other guanine oxidation products and the BER glycosylases that remove them are being uncovered. This work is providing surprising and intriguing new insights into the process of base excision repair.
Introduction
The importance of the specific pairing first postulated by Watson and Crick of adenine with thymine and guanine with cytosine is well known. Indeed, this specificity is key for preserving the informational content of the genome. However, the structural properties of the DNA bases, and therefore the molecular basis for the specific pairing, are often modified via reactions with environmental toxins, such as UV-light and cigarette smoke, as well as endogenous metabolic products or by-products, such as S-adenosylmethionine and hydroxyl radicals.1–3 Over 30 years ago, armed with the knowledge of facile deamination of cytosine to yield uracil, Tomas Lindahl searched for enzymes that mediated repair of uracil and discovered the bacterial enzyme, Uracil-DNA glycosylase (UDG).4, 5 This enzyme catalyzes the removal of uracil in DNA and this reaction is the first step of the base excision repair (BER) pathway (Figure 1).6–8 It is now known that there are many different damage-specific DNA glycosylases that recognize and excise abnormal DNA bases as the first step of BER. Once the base has been removed, other enzymes are recruited to excise the remaining sugar fragments, and reinstall an unadulterated nucleotide to constitute “repair” of the damaged base site.9
Figure 1. Base Excision Repair (BER) Pathway.

Short-patch BER pathway is illustrated in its minimal form.9 A damage-specific glycosylase removes a damaged base to produce an apurinic-apyrimidinic (AP) site within DNA. Further processing by an AP endonuclease and a phosphoribosyl lyase create the proper ends for a DNA polymerase. Using the opposite strand as a guide, the repair polymerase installs the appropriate nucleotide. DNA ligase seals the backbone. Note: many DNA glycosylases (such as hOGG1) harbor an additional AP endonuclease stand scission activity.
In the past few years, surprising insight into these enzymes has come from a variety of disparate directions that have propelled the damage-specific glycosylases into the limelight. Specifically, structural, biophysical and biochemical studies have provided exquisite insight into the remarkable process by which these enzymes locate damaged DNA bases in the context of a huge excess of normal DNA. Genetic studies have revealed direct correlations of defects in the repair of damaged bases with human diseases. Lastly, the use of sophisticated analytical and chemical approaches have resulted in identification of new base lesions and the glycosylase enzymes that act upon them. In this review, we will highlight the base excision repair of guanine oxidation products and illustrate new features of the chemistry, structural biology and clinical aspects of these repair pathways.
The special problem of 8-oxoguanine
Production of reactive oxygen species (ROS), such as hydrogen peroxide, superoxide and hydroxyl radicals, has been linked to the initiation and progression of cancer.10 ROS are produced as by-products of cellular respiration or as part of inflammatory responses. In addition, ROS can be produced as a consequence of ionizing radiation or due to environmental exposure to transition metals, chemical oxidants or free radicals. In normal cells, enzymatic and nonenzymatic mechanisms are present to counterbalance the production of ROS. However, aberrantly functioning cells are often in a state of “oxidative stress” where the balance of oxidants to antioxidants has been disrupted leading to elevated levels of cellular damage.
The bases of DNA are particularly susceptible to oxidation mediated by ROS.11 The low redox potential of guanine makes this base particularly vulnerable and leads to a plethora of potential oxidized guanine products.11 The most thoroughly examined and most famous guanine oxidation product is 7,8-dihydro-8-oxo-2′-deoxyguanosine (OG);11, 12 indeed, the presence of OG is often used as a biomarker in cells to indicate the extent of oxidative stress.10 The OG lesion is particularly sinister due to its modest two atom change from G, an oxo group at the 8-position and a H atom at N7, and its ability to functionally mimic T when in the syn conformation to form a stable OG(syn): A(anti) base pair (Figure 2). In contrast to many other types of DNA damage, these structural features provide for efficient, though inaccurate, bypass of OG by replicative DNA polymerases.13 A series of structures of DNA polymerase I fragment from Bacillus stearothermophilus (BF) illustrated the consequences of OG in the template prior to insertion, after insertion of A or C, and after extension of both OG-containing base pairs.14 A comparison of these structures to those containing mismatches in DNA showed that inaccurate bypass of OG results from the ability of the OG(syn):A(anti) base pair to mimic a normal base pair when processed by BF. In contrast, the creation of OG(anti):C(anti) base pairs during replication induces template and polymerase distortions similar to those observed in the corresponding structures of mismatches created within the polymerase active site. The structural information provide a molecular rationale for the more facile insertion of A than C opposite OG, as well as the lack of error detection after insertion by replicative DNA polymerases.
Figure 2. Structures of Base-pairs containing 8-Oxoguanine.

The subtle change of placing an oxo-group at C8 and an NH at N7 of G to produce 8-oxoguanine (OG) allows for facile base-pairing with both C and A. The structures of the base pairs of OG:C and OG:A are compared to G:C and T:A
Failure to remove OG prior to replication results in G:C to T:A transversion mutations.13, 15 Repair pathways are present in all organisms that are tailored to deal with the special features of OG and mitigate its high mutagenic potential. MutT, MutM (Fpg), and MutY enzymes in bacteria16 and the corresponding human enzymes,8 MTH1, hOGG1 and MUTYH,# constitute the “GO” repair pathway (Figure 3). MutT and MTH1 hydrolyze dOGTP, removing it from the nucleotide pool and preventing incorporation of this damaged nucleotide into DNA by polymerases during replication. MutM and hOGG1 are BER glycosylases that excise OG from OG:C base pairs (bps), and subsequent processing by the other enzymes in the BER pathway restore G:C bps. The MutY and MUTYH glycosylases intercept OG:A bps and remove the inappropriate adenine base. Subsequent processing of the apurinic-apyrimidinic (AP) site, and replication by a repair polymerase provides an opportunity to create a proper OG:C substrate for the appropriate OG glycosylase. Notably, structural studies of the eukaryotic repair polymerase β with OG-containing templates have illustrated that this polymerase active site more easily accommodates OG:C bps than OG:A mismatches.17 This specificity is exactly opposite of the results with the BF polymerase; moreover, this structural information explains the higher propensity of polymerase β to insert dCTP over dATP opposite OG which is required of this repair polymerase to prevent futile cycles of repair and replication.17
Figure 3. Base Excision Repair Pathway for 8-Oxoguanine.

The presence of OG in DNA causes G:C to T:A transversions as illustrated in the center panel. The human BER glycosylases hOGG1 and MutYH remove OG and A from OG:C and OG:A base pairs, respectively. The corresponding enzymes in bacteria are MutM/Fpg and MutY. MutT and MTH1 are not shown, but play an important role in preventing incorportion of OG by hydrolyzing d(OGTP).
Seeking and removing OG lesions
In eukaryotic cells, it has been estimated that there are several OG residues for every 106 guanines within the nuclear DNA.18 This low frequency and the similarity in structure of OG and G, makes the task required of OG glycosylases to find OG lesions among a sea of undamaged bases a formidable challenge. Other BER glycosylases, such as UDG and MutY, also require the ability to distinguish relatively minor perturbations from the normal DNA bases or base pairs. Structural and biophysical studies of many DNA glycosylases bound to their relevant substrate duplexes have revealed common themes in lesion recognition that include enzyme-initiated DNA bending, base pair disruption, extrusion of the damaged nucleotide from the interior of the DNA helix and placement of the target base into a base-specific enzyme pocket.19–23 This enzyme driven process is often referred to as “base-flipping” but may be more accurately described as “nucleotide-flipping” since the entire nucleotide is rotated out of the helix to accommodate the base in the base specific pocket.22 The molecular gymnastics mediated by the damage-specific glycosylases has been described as a routine involving specific “pinch-push-pull-plug” steps.19, 20 Upon encounter of a specific DNA lesion, the damage-specific glycosylase induces bending and distortion of the DNA double helix, which constitutes the “pinch.” This step, as well as a “push” of the target base out of the helix, is facilitated by intercalation of a glycosylase amino acid side chain into the DNA helix. The same or another intercalating amino acid also serves as a “plug” to fill in the position of the extruded base and stabilize the contortion of the DNA duplex. A “pull” by active-site residues specific for the relevant target base secures it into the recognition and clipping pocket.
These general features used for lesion recognition are illustrated well with the structure of a catalytically inactive K249Q variant of hOGG1 bound to a substrate OG:C-containing duplex.24 The K249Q variant retains high affinity binding to an OG:C-containing duplex, but is unable to excise OG due to the mutation of Lys 249 that participates in catalysis of the base-displacement reaction. Such complexes with the substrate are referred to as lesion recognition complexes (LRCs). In the LRC X-ray structure, the OG:C base pair has been disrupted and the OG nucleotide has been rotated to place the attached base within an OG-specific pocket (Figure 4). Though hOGG1 makes many specific contacts to both the extrahelical OG and the orphaned C, only one hydrogen-bonding contact would not be possible with G. This crucial hydrogen bond is between the carbonyl oxygen of hOGG1’s Gly 42 to the N7H of OG. With G, not only would such a hydrogen bond be absent, but there would also be unfavorable repulsion between the carbonyl oxygen and the lone pair at N7. It seems unlikely that specificity for OG over G would be due to a single hydrogen bond and therefore the mechanism for discrimination between these two bases is likely much more complex.
Figure 4. Recognition of OG by hOGG1 observed in the lesion-recognition complexes of hOGG1 with OG:C-containing duplexes.

This is a view of the base specific pocket of hOGG1 and shows residues involved in recognizing OG. Importantly, an OG specific hydrogen-bond between NH7 and the Gly 42 carbonyl provides specificity for OG over G. In addition, the dipole associated with the Lys249/Cys 253 pair makes favorable dipole-dipole interactions with OG, but not G.
Revealing the strategy used by hOGG1 to discriminate between OG:C and G:C bps required a structural glimpse of hOGG1 encountering a G:C bp. Since these enzymes do not recognize G:C bps, such an encounter is a transient process and therefore Verdine and coworkers utilized an innovative covalent trapping method to secure hOGG1 in proximity to a G:C bp.25 Using the structure of the hOGG1 LRC as a guide,24 a cysteine residue within the enzyme was installed and a single thiol-modified base-containing nucleotide was positioned within the DNA duplex such that a disulphide crosslink (DXL) would be formed when hOGG1 encountered the targeted G:C bp. The remarkable structure25 revealed that even though G is forcibly presented to the enzyme, it does not gain access to the OG site, but is lodged within an alternative peripheral “exo” site (Figure 5). Calculations of differences in free energy indicate that favorable OG interactions and unfavorable G interactions in both the active site and the exo site result in the 105-fold preference for OG over G. Quantum calculations also showed that the OG and G nucleobases possess local dipoles that are oriented in opposite directions due to the charge inversion at C8 and N7. The hOGG1 active site possesses a complementary dipole for OG created by a Lys 249-NH3+/Cys 253-S− amino acid pair within the active site (Figure 4). Indeed, the calculated contribution of this dipole-dipole interaction to recognition is greater than the contribution of the single hydrogen bond to NH7 of OG. However, the importance of the specific hydrogen bond to OG was illustrated by replacement of Gly 42 with alanine and structural analysis of the resulting complexes using the DXL approach with OG or G presented to the enzyme.26 Surprisingly, in all of the structures examined, the backbone conformation at position 42 remains identical to that in the LRC (Figure 4) despite the fact that the alanine substitution is energetically unfavorable due to the steric demands to the side chain methyl group. These structures show the role of the surrounding protein in “hard-wiring” the conformation of Gly 42 to preserve the ability of hOGG1 to recognize OG, and effectively reject G from the active site.26
Figure 5. Comparison of LRC of hOGG1 to nonspecific complexes examining G:C bps observed using the DXL approach.

In all structures, the hOGG1 backbone is represented as a dark grey ribbon while the DNA is in gold. Important amino acid side chains are colored fuchsia (Asn or Cys 149), blue (Tyr 203), aqua (Arg 204), purple (Arg 154), orange (Gly 42), light grey (Phe 319), lime (His 270), and pink (Gln 315). Panel A: hOGG1 LRC (with OG:C-containing DNA) where OG is shown in red and the estranged C in green. Panel B: hOGG1 interrogating a G:C bp, with the target G in hot pink and target C in green (Arg 154 is not shown). Panel C: hOGG1 interrogating a G:C bp adjacent to an OG lesion, with OG in red, the target G in hot pink, and the target C in green.
The presence of the exo base binding site and the calculations showing preferences for binding OG at both base binding sites, suggest that the damaged base is extracted by hOGG1 from the duplex in discrete steps; an initial step being placement into the exo site and a final step being engagement into the active site pocket (Figure 5). This work also reveals a strategy for accurate damage recognition. The use of a multi-step process of base excision allows for many opportunities to check the identity of the base prior to its excision. In this case with hOGG1, only OG has the proper credentials to make it through all of the checkpoints. A later intermediate in the OG recognition and excision process was recently revealed by structural analysis of several constructs which contained a mutation that enlarged a side chain (Q315F) (Figure 4) to occlude the active site pocket.26 Notably, in one structure, the OG lesion is almost, but not quite, completely inserted into the active site pocket; however, the OG base is not clipped out of the DNA with this mutated enzyme. This illustrates another level of quality control that requires perfect geometric alignment of the base within the specificity pocket to access the proper transition state for base cleavage.
The DXL approach also provided a fortuitous structure of hOGG1 interrogating a G:C bp within a DNA duplex located next door to an OG:C bp (Figure 5).27 This structure reveals how OG may initially be detected by hOGG1 during the evaluation of the neighboring base pair. The overall structure is very similar to the LRCs and the interrogation complex with the G:C bp discussed above; however, though the target G is destacked from the helix, it does not reside at the exo site. Rather, the target G is folded back into the major groove and forms an unusual base-triple hydrogen bonding interaction with the major groove face of the neighboring OG. The altered mode of examination of G in the presence of OG appears to be related to severely repulsive steric and electronic clashes with the phosphate backbone due to the presence of the 8-oxo group of the OG residue. This results in altered backbone conformations that do not allow for complete presentation of the G to the exo-site and thus favor the folded-in conformation that is further stabilized by hydrogen-bonding with OG. The authors also point out that such base-phosphate clashes only occur due to the remodeling of the duplex by hOGG1. Thus, the active role of the glycosylase in probing and altering the duplex is a key feature of the mechanism for detection of subtle alterations of the base structure.
The structures above suggested that G:C base pairs may require special attention by hOGG1, but does hOGG1 scrutinize all normal base pairs by removing one of the bases from the helix? Such a process would seem to be energetically costly and inefficient. To further explore the interrogation process used by OG glycosylases, the DXL approach was used to generate several structures of Bacillus stearothermophilus MutM, which is functionally similar to hOGG1, tethered to normal DNA in proximity to both an A:T and G:C bp (Figure 6).28 In these structures, the normal bases are not extruded from the helix suggesting that MutM initially locates OG lesions by intrahelical interrogation. Indeed, the side chain of a phenylalanine residue (Phe 114) was found to be wedged into the helix from the minor groove above an A:T or G:C bp. This invasion by Phe 114 results in severe bending of the DNA and buckling of the base pair. In addition, two other amino acid residues (Arg 112 and Met 77) that take the place of the flipped-out OG base in the corresponding LRC29 are drawn back, as if waiting to lunge-in when space has been created. The overall structure of these nonspecific complexes are similar to that of the LRC, except that there are many water-mediated contacts rather than direct contacts between the enzyme and DNA. This may allow for a looser association with the DNA to facilitate movement along the helix. Interestingly, in using the DXL strategy with MutM, it was difficult to trap a complex in which MutM was interrogating a G:C bp; indeed, the protein appeared to avoid interrogating G:C bps. The desired structure of interrogation of a G:C bp required changing of the DNA sequence to present only a string of G:C bps to the enzyme. The various structures taken together suggest that Phe 114 serves as a sensor of the stability and/or deformability of the target base pair. Notably, OG:C bps are slightly less stable than A:T bps; however, they are considerably less stable than G:C bps. Moreover, the non-standard properties of an OG:C bp may not withstand the probing by Phe 114 of MutM. Once the base pair is disrupted, the liberated OG would then be captured by the active site of MutM.
Figure 6. Comparison of LRC of MutM to interrogation complexes with G:C and A:T bps observed using the DXL approach.

In all structures, the MutM backbone is represented as a dark grey ribbon while the DNA is in gold. The intercalating Phe 114 is shown in fuchsia, while Arg 112 and Met 77 are in blue and aqua, respectively. Panel A: MutM LRC with OG in red and estranged C in green. Panel B: Interrogation of a G:C bp by MutM, with the target G in red and target C in green. Panel C: Interrogation of an A:T bp by MutM, with the target A in red and target T in green.
One might expect that such a search process would not be extremely precise and therefore Banerjee et al.28 suggest that the glycosylases may make-up for this imprecision by being extremely fast to allow for multiple opportunities to find a damaged base. This fast search process of hOGG1 was recently visualized using single-molecule detection to track hOGG1 movements along a normal DNA duplex.30 In these studies, at physiological salt and pH, hOGG1 was found to track along the DNA with a diffusion constant of D = 1.2 × 106 bp2/s. Based on this nearly diffusion-limited value, the authors estimate that the barrier for sliding must be extremely small (0.5 kcal/mol). This was consistent with the observed unbiased random movement of hOGG1 on DNA, and suggests that hOGG1 rapidly searches along the helix due to Brownian motion. Mutation of His-270 located at the DNA-binding interface,24–26 together with pH studies, suggests that the DNA-protein interface has been optimized to provide for fast enzyme sliding.
The single-molecule studies of hOGG1, along with the many structures of trapped complexes of hOGG1 and MutM, provide for a scenario of events involved in the search for OG lesions by hOGG1 or MutM to be envisioned (Figure 7). First, the OG glycosylase moves rapidly along the helix inserting the probe ligand into the helix looking for vulnerable sites. Intercalation of an amino acid residue of the enzyme at a normal base pair merely buckles the base pair; however, one could imagine that such a probing event may disrupt an abnormal base pair like OG:C. Such a search process would be extremely fast such that sometimes an OG base may be missed, but this may be preferable to wasting time interrogating undamaged DNA. This fast, random and redundant process would provide many opportunities to find OG. Once the OG has been partially expelled from the helix, it would be captured by the exo-site of hOGG1 and quickly passed to the OG-specific active site. Occasionally, a G residue may be displaced from the helix, however, this is intercepted in the exo-base binding site, and does not proceed to the OG-specific pocket, but instead is replaced back within the helix. Moreover, if a G residue adjacent to OG is interrogated, it does not fully dislodge from the helix and thus allows the OG glycosylase to more rapidly hone-in on the adjacent OG lesion. Structural and biophysical studies suggest that similar mechanisms are likely operative with other glycosylases.20, 31 Indeed, some damaged bases may be more readily expelled from the helix or be more readily distinguished from their normal counterpart making such a search process more streamlined.
Figure 7. Finding OG lesions- schematic representation of search process based on structures of hOGG1 and MutM trapped complexes.

The enzyme tracks rapidly along DNA, inserting a “probe” amino acid residue (shown as a green hexagon) at various base-pairs to test the stability/deformability of the duplex. This results in preferential expulsion of OG from OG:C bps. However, if a G from a G:C bp is extruded, it is captured by an external site and then replaced within the helix. Encountering a G:C bp adjacent to an OG:C bp allows for facile detection of OG since this G cannot be extruded in the same fashion, and will then promote movement of the enzyme to the OG:C bp. At this point, the OG may first be extruded to the exo-site and then is quickly captured in the OG-specific pocket to be clipped-out of the DNA.
The partners for MutM and hOGG1 in the GO repair pathway, MutY and MUTYH also require precise OG recognition to selectively remove only adenines harbored in OG:A mismatches. In fact, MutY has a special C-terminal domain not found in other BER superfamily glycosylases, that plays an important role in recognition of OG.32–34 In the crystal structure of an inactive variant of Bacillus stearothermophilus MutY (D144N BsMY),34 there are a plethora of contacts to OG, but minimal contacts to adenine. Despite the implied importance of OG, MutY also mediates removal of adenine opposite other bases, including G, FapydG and C.6, 35 In fact, MutY was originally discovered as an enzyme capable of repair of G:A and C:A bps.36–39 Time-resolved fluorescence experiments of the MutY adenine excision reaction with OG:A substrates indicated a multi-phase reaction profile with a fast process associated with changes at OG, and a slower process associated with altering the environment of the adenine base.40 The fluorescence data was originally interpreted as resulting from sequential extrusion of OG, then A from the helix. However, in the BsMY LRC structure,34 the OG is within the helix but with an altered conformation about the N-glycosidic bond. Though the molecular explanation for the OG-related fluorescence changes is not clear, together with the structure, these experiments suggest that MutY relies heavily on initial recognition of OG to locate adenine bases for excision. Additional structures of MutY bound to other base pairs (such as T:A) would be of considerable interest to elaborate how this enzyme prevents inadvertent excision of adenine opposite thymine.
Base Excision Repair and Colorectal Cancer
The importance of preventing mutations associated with 8-oxoguanine was highlighted when a direct link between mutations in the gene encoding the human homologue of MutY (MUTYH) and colorectal cancer was uncovered.41 This work also established the first link between inherited defects in base excision repair and cancer. In a British family (Family N), several siblings presented clinical symptoms characteristic of Familial Adenomatous Polyposis (FAP), a common form of familial colon cancer. In FAP, the colon of afflicted individuals is littered with adenomatous polyps due to mutations in the adenomatous polyposis coli (APC) gene.42 The APC protein plays many important roles in controlling growth of colon cells, and is mutated in the majority of colorectal tumors. The familial nature of FAP usually results from inherited mutations in APC, however, this was not the case in Family N. Consistent with the polyposis phenotype, tumors from afflicted individuals of Family N possessed inactivating somatic APC mutations, but with an unusually high level of G:C →T:A transversions. Since this type of mutation is commonly associated with OG, the genes encoding the human counterparts of the “GO” repair pathway of the afflicted family members were sequenced. This revealed germline biallelic missense mutations in MUTYH that would produce two variants of MUTYH, each containing a single amino acid substitution, specifically Y165C and G382D MUTYH. Due to the high similarity of MUTYH and E. coli MutY, the activities of the two corresponding variants in E. coli MutY were analyzed.41 Both were found to have compromised adenine removal activity relative to WT E. coli MutY. This supported the hypothesis that the corresponding MUTYH variants have a reduced capacity to initiate the repair OG:A mismatches, which leads to increased levels of G:C → T:A mutations in APC, eventually resulting in inactivation of the APC protein. This is a novel mechanism by which inherited defects in a gene for a BER enzyme (MUTYH) lead to mutations in another cancer predisposing gene (APC). This new colorectal cancer predisposition mechanism is now referred to as MUTYH-associated polyposis (MAP).43
In order to evaluate the consequences of the two amino acid variations on damage/mismatch recognition separately from base cleavage, dissociation constants of the two variant MutY enzymes were measured with DNA duplexes containing a noncleavable A analogue, 2′-deoxy-2′-fluoroadenosine (FA), opposite OG and G.44 In these experiments, Y82C and G253D MutY exhibited reduced affinity for both OG:FA- and G:FA-containing duplexes relative to the WT enzyme. Surprisingly, the duplex affinity of the variants was not enhanced by the presence of OG, as is the case with the WT enzyme, indicating that the mutations abrogate the ability to recognize OG over G. Insight into the role that these two amino acids play in OG recognition was also illustrated in the structure of D144N BsMY bound to an OG:A-containing duplex (Figure 8).34 The corresponding Tyr in BsMY (Tyr 88) was found to be intercalated 5′ to OG and participating in a hydrogen-bond network with the NH7 of OG, while the Gly residue (Gly 260) is located in a turn region of the protein where backbone amides are involved in intimate hydrogen-bonds with the phosphodiester backbone adjacent to OG.
Figure 8. Structure of D144N BsMY bound to an OG:A Mismatch-containing duplex highlighting corresponding amino acid variations observed in MAP.
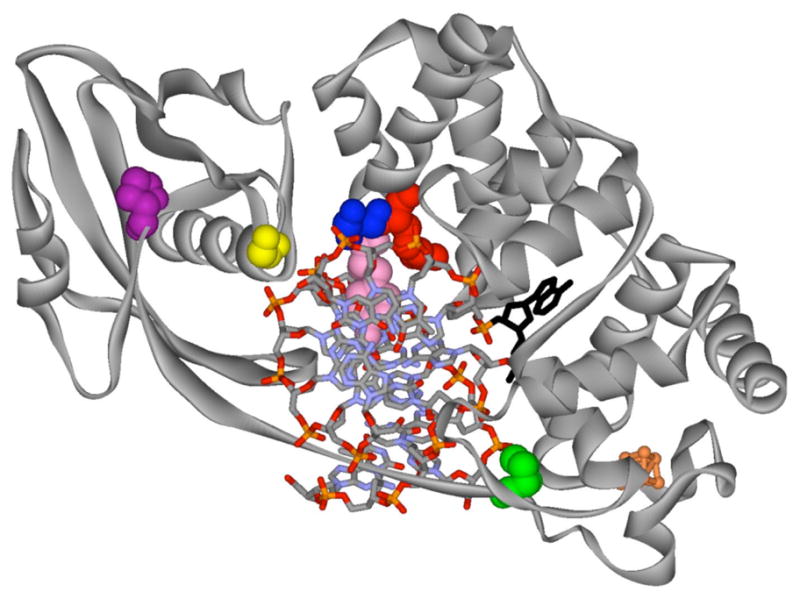
The view is down the helix axis with BsMY encircling the DNA. The flipped-out A is shown in black, while the 4Fe-4S cluster of BsMY is shown in orange. Residues in BsMY and corresponding variations in MUTYH are as follows: Y88 (pink), Y165C; G260 (yellow), G382D; S90 (blue), S167F; R91 (red), R168L; P226 (green), P345T; P269 (purple), P391L.
Interestingly, the location of Tyr 82 in MutY corresponds to the Phe 114 “probe” amino acid revealed in the structure of MutM trapped with nonspecific DNA.28 This suggests that the ability to seek and find OG:A mismatches by MutY is hampered by mutation of the large Tyr probe ligand to a smaller cysteine residue. The importance of a bulky residue for intercalation to indirectly recognize OG:A mismatches was demonstrated by the observation that the Tyr residue may be replaced by Leu without sacrificing catalytic activity.45 A leucine side-chain is also present at the analogous position in many other glycosylases,25 but should not be able to participate in the hydrogen-bonding network to NH7 of OG. In a more subtle fashion, the turn region of BsMY containing Gly 260 appears to be stabilizing a “pinched” and distorted conformation of the phosphate backbone adjacent to OG that may be required to promote adenine extrusion. This Gly residue is located within the unique C-terminal domain of MutY that has been shown to be important for OG recognition.32–34 Interestingly, the single amino acid variation in G253D MutY provided a loss in affinity for duplexes containing OG over G that was similar to that observed with a truncated form of MutY completely lacking the C-terminal domain.44 Inspection of the D144N BsMY-OG:A mismatch DNA structure34 revealed that the peptide backbone torsion angles of glycine in this turn region are highly unusual, and would only be easily accommodated by the flexible glycine. Thus, replacement of this glycine with Asp or Ala destabilizes the turn region and alters the ability to specifically recognize OG at this site.45
Though both variant MutY enzymes have a DNA binding defect, the binding defect of Y82C in vitro translates to a more deleterious effect on adenine excision.44, 45 In vitro adenine glycosylase activity of Y165C and G382D MUTYH46 and the corresponding variants in the murine MutY enzyme (mMutY##)47 were analogous to those observed with the E. coli variants. However, in contrast to the E. coli variants, the activity of mMutY variants is further reduced in the presence of other DNA binding proteins.47, 48 For example, the presence of the human AP endonuclease (Ape1) enhances the adenine excision activity of WT mMutY by stimulating release of mMutY from the AP-site DNA product. In contrast, with the mMutY variants, the presence of the Ape1 reduces the efficiency of the adenine excision activity.47 This is a consequence of competition between MutY and Ape1 for the OG:A-containing DNA substrate, which is not a problem for the WT enzyme. These competition effects may also explain the fact that both human variants in cell extract preparations appeared completely inactive.49 Taken together, these observations suggest that defects in finding and recognizing damaged DNA may be magnified in a cellular context compared to in vitro experiments due to the presence of many other DNA binding enzymes. In addition, the search process is much more demanding in a cellular environment due to the larger size of the DNA molecules (kilobases) compared to the small duplex substrates used in vitro (30 bps). These ideas may explain why both mMutY and MUTYH variants also appear less active in cellular complementation assays.44, 50
Consistent with a global “GO” BER defect, a high proportion of tumors from patients with biallelic MUTYH mutations have also been observed to harbor G to T transversion mutations in the first G of codon 12 of the K-ras oncogene.51 Thus, this suggests that other genes, in addition to APC may be susceptible to mutation due to dysfunctional MUTYH; however, to date no other compelling connections with other types of cancer have been reported. The mutation of K-ras in MAP tumors was also interesting because the same type of inactivating mutation is observed in mice deficient in both mMutY and mOgg1.52 The double knockout mice exhibited a high susceptibility to tumor formation, predominantly lung and ovarian tumors, and lymphomas. In addition, these mice also exhibited increased levels of OG in the lung and small intestine.53 Surprisingly, mice deficient in only mMutY do not exhibit any atypical properties.52 However, crossing of mMutY-deficient mice with multiple-intestinal neoplasia (ApcMin/+) mice, which carry a nonsense mutation in APC, resulted in increased intestinal tumorgenesis, compared to ApcMin/+/mMutY+/+ mice.54 These mouse model studies showed that mMutY deficiency does result in an increased intrinsic mutation rate, however, this alone does not lead to tumor formation in mice. Deficiencies of other proteins, such as APC or Ogg1, aid in fueling the progression towards cancer. In humans, the extent of oxidative stress and level of mutations mediated by OG, especially in the intestinal tract, may be greater than in mice. It should also be noted that in the mouse models, the mMutY protein is absent, rather than present in a variant form. Studies of mMutY variant knock-in mice would be quite interesting since this would illuminate whether the presence of the variant mMutY proteins may enhance mutagenesis, and thus be more deleterious than the complete absence of the protein.
Beyond Family N
Since the original discovery of Family N, considerable work has established the relationship between mutations in MUTYH and colorectal adenomas and carcinomas and this disorder is now referred to as MAP.43, 55–60 MAP appears to be an autosomal recessive disorder requiring mutations in both alleles. Present estimates of the frequency suggest that MAP accounts for approximately 1% of all colorectal cancer (CRC); though this number may increase as more patients are tested for mutations in MUTYH.61 This contribution to CRC is somewhat less than present estimates for hereditary nonpolyposis colorectal cancer (HNPCC) of 1–6%.62, 63 HNPCC, like MAP, is due to an inherited DNA repair defect, specifically in the mismatch repair (MMR) genes.63 Of the MMR gene mutations, those in the genes encoding the human MutS homologue, hMSH2 and the human MutL homologue, hMLH1 predominate.63 However, HNPCC is distinctly different from MAP in being an autosomal dominant disorder and in resulting from a distinctly different type of DNA repair defect (MMR vs BER). Thus, the molecular evolution leading to cancer originating from these two repair defects is not expected to be exactly the same. Though FAP and HNPCC are relatively rare, the understanding of the genetic basis of these disorders has provided critical insight into pathways leading to cancer.64, 65 For example, a feature of the MMR defect is the expansion or contraction of short nucleotide repeats in DNA of tumors which is referred to as microsatellite instability (MSI).62, 63 Interestingly, MSI is also characteristic of many sporadic colorectal cancers.66 The presence of MSI, family history, the presence and type of MMR gene mutation, and clinical manifestations are all important pieces of the puzzle that physicians and genetic counselors use to diagnose HNPCC and appropriately advise and treat patients and their families.67 Though much is known about HNPCC, many questions still remain about the molecular mechanism leading to cancer that starts with an MMR defect.
Since MAP has only recently been discovered, more information regarding the clinical and molecular properties of MAP is needed to aid in diagnosis and treatment of affected patients and their family members. Such knowledge may also provide insight into how MUTYH mutations contribute more globally to cancer. Presently, testing for mutations in MUTYH is recommended for patients exhibiting clinical features of FAP that either have been shown to not have inherited mutations in APC or do not have a family history consistent with dominant transmission of FAP as is characteristic of APC mutations.56, 57 In classical FAP, the sheer number of adenomatous polyps leads to a lifetime risk of eventual carcinoma approaching 100%.42 Based on the preliminary studies,59, 60, 68, 69 a similar lifetime risk for colorectal cancer has been suggested for MAP.63 The potential increased colorectal cancer risk for heterozygotes harboring only one mutated MUTYH allele has been a subject of debate, and will likely require more clinical testing to be resolved.70
In addition to the two variants in MUTYH that were initially identified (Y165C and G382D), ~82 different germline mutations have been found in MUTYH of patients with colorectal adenomas and carcinomas (Figure 9).70 The two variants, Y165C and G382D appear to be the most common mutations found in caucasians of Northern European origin.43, 70 Some mutations have been found to be prevalent in certain ethnic populations.70–72 Though many of these mutations may lead to lack of MUTYH protein, there are a large number of missense mutations (~52) that present a diagnostic challenge since their significance is unknown. On the basis of the type of mutation, the location in the sequence of MUTYH, and the corresponding position in the structure of BsMY-DNA, it is possible in some cases to make a prediction of the possible consequence (Figure 8). For example, early truncating mutations (e.g. Y90X) or late truncating mutations (E466X) in the core of the C-terminal domain are likely to be destablizing and result in the absence of MUTYH protein. With some of the missense variants that are conserved in BsMY, it is possible to suggest that they will affect DNA binding (e.g. R168L, S167F) or affect folding of one of the domains (e.g. W117R). However, the consequence of some conserved missense variants (e.g. P325T, P391L) is not neccessarily obvious. Moreover, some missense variants are not conserved in the bacterial MutYs, and therefore it is difficult to predict an outcome. At this early stage of the understanding of MAP, it is not possible to strictly rule out that some mutations are nonpathogenic polymorphisms that are somehow correlated with patients with colorectal adenomas and carcinomas. It is also possible that some variants are only mildly pathogenic and this may be correlated to the severity of the disease. Indeed, the in vitro results suggest that G382D MUTYH is less catalytically compromised than Y165C MUTYH; however, thus far it is not clear how this correlates with the clinical data. Moreover, besides Y165C and G382D MUTYH, the adenine removal activity has only been examined with four other variants (R227W, V232F, R231L, A459D).73–75 The low frequency and uncertain pathogenicity of this large number of new variants is a considerable problem for genetic counseling of patients. Additional clinical and functional data will be particularly important in revealing any special relationships between specific mutations in MUTYH and colorectal cancer.
Figure 9. Germline Mutations Observed in MUTYH of MUTYH-associated Polyposis.

Shown is an alignment of E. coli MutY, Bacillus stearothermophilus MutY (BsMY) and human MutY homologue (MUTYH). Examples of some of the mutations that have been observed in MAP are indicated. Critical DNA binding motifs are the helix-hairpin-helix (HhH) and the iron-sulfur cluster loop (FCL).110, 111
Why have inherited mutations in MUTYH been correlated with cancer and not other BER enzymes? It is possible that dysfunction of MUTYH may be particularly problematic since there are no other mechanisms for repairing OG:A mismatches. In contrast, nucleotide excision repair or transcription-coupled nucleotide excision repair appear to serve as a “backup” repair mechanism to mediate removal of OG in mouse cells lacking mOgg1.76–79 However, it is also important to note that the clinical feature of colonic polyps provides a key piece of information that prompts APC or MUTYH testing. This narrowed the field of patients tested and increased the probablity of detecting a relatively rare defective gene. Thus, we predict that dysfunction of other BER glycosylases will also be found to be modifiers for cancer susceptibility and these correlations may be uncovered as screening for gene mutations progresses. Moreover, as more information on the molecular basis for cancer becomes available additional links between BER defects and cancer will be more readily revealed.
Beyond 8-oxoguanine
Though 8-oxoguanine has garnered much attention, a cadre of oxidized guanine lesions have been identified (Figure 10).11 Numerous studies indicate that the most common mutations observed from cells under conditions of oxidative stress are G → T and G → C transversions.11 While G → T transversions may be readily explained by the presence of OG or FapydG, other oxidized lesions are likely responsible for mediating G → C transversions. Of these other lesions, the hydantoin products, spiroiminodihydantoin (Sp) and 5-guanidinohydantoin (Gh), have a particularly interesting history in their identification, and are emerging as important lesions to be reckoned with by repair enzymes. For many years, the product of singlet oxygen damage to G was thought to be 4-hydroxy-8-oxo-7,8-dihydroguanosine (4-OH-OG)80, 81 and the presence of this product was used routinely as an indicator of singlet-oxygen damage.82 However, Burrows and co-workers determined the structure of Sp, which coincidentally had the same molecular weight as 4-OH-OG, as a major product formed by further oxidation of OG-containing nucleosides and oligonucleotides.83–86 It was later confirmed that the structure of 4-OH-OG had been mis-assigned and that 4-OH-OG was actually Sp.87, 88 Once the identity of Sp was unmasked, this product has been observed to arise from either G or OG oxidation with a large number of oxidants including singlet oxygen, high valent metal ions, peroxynitrite and ionizing radiation.11, 86 Interestingly, the studies of oxidation of OG revealed that, depending on the conditions, Gh is formed in addition to Sp.84 The predominant product in nucleosides or ss DNA is Sp, while Gh predominates in ds DNA.89
Figure 10. Guanine oxidation products discussed in this review.

A large number of guanine oxidation products have been observed,11 however, in this review we focus on the hydantoin lesions, guanidinohydantoin (Gh) and spiroiminodihydantoin (Sp). The structures of these lesions resemble OG and FapydG in retaining hydrogen-bonding functionality (red) that can mimic T.
The hydantoin lesions have also been demonstrated to be highly mutagenic by both in vitro and in vivo data. Single nucleotide insertion and primer extension experiments using E. coli Klenow fragment of DNA polymerase (KF exo−) indicate that dAMP and dGMP are inserted opposite these oxidized lesions.90, 91 In an E. coli based mutagenesis assay with single-stranded lesion containing viral DNA, Henderson et al. showed that Gh is bypassed as efficiently as OG, while Sp is a strong block to replication.92 Moreover, Gh causes exclusively G→C transversions while the presence of Sp results in a mixture of G→ T and G→ C transversion mutations. Importantly, the mutation frequency for Gh and Sp was nearly 100%, while OG is only mildly mutagenic in these assays (3%).92
An important feature that influences the mutagenic potential of a given lesion is the efficiency of its repair, and the sensitivity of the repair enzymes to the correct base-pairing context. The relatively low mutation frequency of OG, in both E. coli and mammalian cells, is due to the efficient repair of OG. In vitro assays have shown that Gh and Sp are substrates for E. coli Fpg (MutM).89 Efficient repair in the correct base-pairing context would be expected to mitigate the mutagenic potential of lesions, however, removal in the wrong context may actually enhance mutagenesis. This is illustrated with Gh which was found to be excised by Fpg from Gh:G and Gh:C bps with similar efficiencies. The Gh:G bp is particularly interesting since this bp may mediate the G → C transversions observed in vivo. Removal of Gh or Sp opposite A by Fpg was reduced relative to opposite C; however, the extent of removal of these oxidized lesions is greater than removal of OG opposite A.89 The activity in base pairing contexts involving A may also be more problematic since MutY was found to be unable to remove A from Gh/Sp:A bps.89
Surprisingly, Gh and Sp are not substrates for hOGG1, though they are removed by the yeast OG glycosylases, yOGG1 and yOGG2.93 Another bacterial glycosylase, Endonuclease VIII (Nei) has also been shown to be capable of removing Gh and Sp.94 Interestingly, this enzyme usually targets oxidized pyrimidines, such as thymine glycol and 5-hydroxycytosine.95 In fact, Gh and Sp, like OG, possess a thymine-like Watson-Crick hydrogen bonding face (Figure 10). Based on the idea that repair may thwart the ability to detect lesions that are present at low levels in cells, Sugden and co-workers used various repair-deficient cells to identify the products formed by chromate oxidation.96 Indeed, Sp was detected by mass spectrometric techniques in chromate-treated E. coli cells deficient in the base excision repair glycosylase Nei.
Recently, mammalian orthologs of E. coli Fpg and Nei were identified and designated as the Nei-like or ‘NEIL’ family of enzymes (NEIL1, NEIL2 and NEIL3).97–100 The crystal structure of NEIL1 revealed an overall structure that is very similar to E. coli Nei and Fpg, though it contains an unusual “zincless” finger motif that is required for its glycosylase activity.101 The substrate specificity of NEIL1 seems to be more similar to that of E. coli Nei than Fpg.97, 102 Reported substrates include 5-hydroxyuracil, formamidopyrimidines and thymine glycol, while the activity for 8-oxoguanine is minimal. In contrast to hOGG1, hNEIL1 also operates on single-stranded and bubble-structured substrates, thus prompting the suggestion of involvement in replication and/or transcription-coupled repair.103 Qualitative reports also showed that Gh and Sp lesions are removed by the murine counterparts of Nei, mNEIL1 and mNEIL2.104 Interestingly, quantitative examination of substrate specificity with hNEIL1 reveal Gh and Sp lesions as the best substrates for these enzymes identified so far.105 Notably, the murine and human NEIL1 have been found to remove Gh and Sp from all base-pairing contexts which would be mutagenic, except when paired with C. The detection of Sp lesions in cells, coupled with the high mutagenic potential of both Gh and Sp and the potentially muddled processes of repair suggests that these lesions will begin to capture more and more of the attention that is presently directed toward OG.
The importance of hNEIL1 in response to oxidative stress has been illustrated by the observation of increased levels of hNEIL1 mRNA in response to ROS.106 Moreover, knockdown of hNEIL1 using RNA interference produces cells that are extremely sensitive to ionizing radiation.107 In addition, an intriguing report also correlated inactivating mutations in hNEIL1 with human gastric cancer.108 These various biological consequences are not surprising for an enzyme involved in repair of oxidative damage, however, some peculiarities with regards to the biological role of hNEIL1 were revealed with the NEIL1 knockout mouse that was recently created.109 In the absence of oxidative stress, both the NEIL1 knockout (nei1−/−) and the heterozygote (neil1+/−) mice exhibit symptoms consistent with metabolic syndrome in humans.109 These symptoms include severe obesity, dyslipidemia and fatty liver disease. It seems somewhat odd for a defect in a BER glycosylase to be correlated with a metabolic disorder; however, these types of disorders have been linked to oxidative stress. The authors suggest that disruption of energy homeostasis consistent with metabolic disorders may be due to the extensive amount of mitochondrial DNA damage observed in the hNEIL1 deficient mice.109 Alternatively, damage to nuclear DNA in specific cell types, such as liver and pancreas may be extensive in the absence of hNEIL, leading to symptoms of metabolic syndrome. Clearly, these interesting results warrant further study as well as illustrate the important role of base excision repair in the overall well-being of organisms.
Concluding Remarks
Since the discovery of base excision repair over 30 years ago, the field has clearly taken its place in the main stream of DNA repair. The plethora of structural information has provided amazing insight into the processes of damage recognition and excision by these enzymes. This structural work will likely be complemented in the future by new biophysical and biochemical approaches that will allow for direct visualization of the search processes by a variety of DNA glycosylases. This may allow for additional intermediates to be observed and provide a more generalized model for damage recognition by these enzymes.
Providing the molecular details of events involved in the processing of damaged DNA bases by DNA glycosylases will be important for further understanding of how the aberrant function of these enzymes is related to disease. Herein, this relationship was illustrated with MUTYH variants where the biochemical and structural information are consistent with a reduced efficiency to recognize and repair OG:A mismatches. This provides a molecular basis for the observed G →T transversion mutations in APC, leading to MAP. However, understanding of MAP is still at any early state and many unanswered questions about the role of the variant enzymes in initiating the process of carcinogenesis remain. For example, does severity of the disease correlate with the type of MUTYH mutation? The intriguing metabolic disorder phenotype of the NEIL1 knockout mice also suggests that interesting relationships between BER and human disease will continue to be uncovered. Further elucidation of the properties of DNA glycosylases and their roles in prevention of mutations associated with altered DNA bases may lead to potential new approaches and targets for manipulation in cell biology and in medicine.
Acknowledgments
We thank members of the David laboratory for reading the manuscript. We also sincerely apologize to all scientists whose original studies and reviews were not included due to space limitations. The research in the laboratory of S.S.D. is funded by the National Institutes of Health. V.L.O. has been supported by pre-doctoral fellowships from the National Institutes of Health (GM08537 and CA093247).
Footnotes
The MUTYH gene is also commonly called MYH or hMYH. However, due to confusion with the gene encoding myosin-heavy chain, it has been suggested that the MUTYH terminology should be adopted (Sampson 2005).
The murine MutY homologue is usually abbreviated as mMYH or Myh. In order to reduce confusion and be consistent and distinct from the abbreviation of the human homologue MUTYH, we are using mMutY.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interest.
Literature Cited
- 1.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 2.Friedberg EC. DNA Damage and Repair. Nature. 2003;421:436–440. doi: 10.1038/nature01408. [DOI] [PubMed] [Google Scholar]
- 3.Pfeifer GP, et al. Tobacoo smoke carcinogens, DNA Damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21:7435–51. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- 4.Friedberg EC. Inroads into base excision repair II. The discovery of the DNA glycosylases. DNA Repair. 2004;3:1531–1536. doi: 10.1016/j.dnarep.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 5.Lindahl T. An N-Glycosidase from Escherichia coli that releases Free uracil from DNA containing deaminated cytosine residues. Proc Natl Acad Sci USA. 1974;71:3649–3653. doi: 10.1073/pnas.71.9.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.David SS, Williams SD. Chemistry of Glycosylases and Endonucleases Involved In Base-Excision Repair. Chem Rev. 1998;98:1221–1261. doi: 10.1021/cr980321h. [DOI] [PubMed] [Google Scholar]
- 7.Fromme JC, Verdine GL. Base Excision Repair. Adv Prot Chem. 2004;69:1–41. doi: 10.1016/S0065-3233(04)69001-2. [DOI] [PubMed] [Google Scholar]
- 8.Barnes DE, Lindahl T. Repair and Genetic Consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445–476. doi: 10.1146/annurev.genet.38.072902.092448. [DOI] [PubMed] [Google Scholar]
- 9.Sung JS, Demple B. Roles of base excision repair subpathways in correcting oxidized abasic sites in DNA. FEBS J. 2006;273:1620–1629. doi: 10.1111/j.1742-4658.2006.05192.x. [DOI] [PubMed] [Google Scholar]
- 10.Klaunig JE, Kamendulis LM. The Role of Oxidative Stress in Carcinogenesis. Annu Rev Pharmacol Toxicol. 2004;44:239–67. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 11.Neeley WL, Essigmann JM. Mechanisms of Formation, Genotoxicity, and Mutation of Guanine Oxidation Products. Chem Res Toxicol. 2006;19:491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 12.Burrows CM, Muller J. Oxidative Nucleobase Modifications Leading to Stand Scission. Chem Reviews. 1998 doi: 10.1021/cr960421s. [DOI] [PubMed] [Google Scholar]
- 13.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 14.Hsu GW, Ober M, Carell T, Beese LS. Error-prone replication of oxidatively damaged DNA by a high-fidelity DNA polymerase. Nature. 2004;431:217–221. doi: 10.1038/nature02908. [DOI] [PubMed] [Google Scholar]
- 15.Wood ML, Esteve A, Morningstar ML, Kuziemko GM, Essigmann JM. Genetic effects of oxidative DNA damage: Comparative mutagenesis of 7,8-dihydro-8-oxoguanine and 7,8-dihydro8-oxoadenine in Escherichia coli. Nucleic Acid Res. 1992;20:6023–6032. doi: 10.1093/nar/20.22.6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Michaels ML, Miller JH. The GO system Protects Organisms from the Mutagenic Effect of the Spontaneous Lesion 8-hydroxyguanine (7,8-dihydro-8-oxoguanine) J Bact. 1992;174:6321–6325. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krahn JM, Beard WA, Miller H, Grollman AP, Wilson SH. Structure of DNA polymerase B with the Mutagenic DNA Lesion 8-oxodeoxyguanine Reveals Structural Insights into its Coding Potential. EMBO J. 2003;11:121–127. doi: 10.1016/s0969-2126(02)00930-9. [DOI] [PubMed] [Google Scholar]
- 18.Gedik CM, Collins A. Establishing the background level of base oxidation in human lymphocyte DNA: results on an interlaboratory validation study. FASEB J. 2005;19:82–84. doi: 10.1096/fj.04-1767fje. [DOI] [PubMed] [Google Scholar]
- 19.Parikh SS, Putnam CD, Tainer JA. Lessons learned from structural results on uracil-DNA glycosylase. Mutat Res. 2000;30:183–199. doi: 10.1016/s0921-8777(00)00026-4. [DOI] [PubMed] [Google Scholar]
- 20.Stivers JT. Site-Specific DNA Damage Recognition by Enzyme-Induced Base Flipping. Prog Nucl Acid Res Mol Biol. 2004;77:37–65. doi: 10.1016/S0079-6603(04)77002-6. [DOI] [PubMed] [Google Scholar]
- 21.Fromme JC, Banerjee A, Verdine GL. DNA glycosylase recognition and catalysis. Curr Opin Struct Biol. 2004;14:43–49. doi: 10.1016/j.sbi.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Huffman JL, Sundheim O, Tainer JA. DNA base damage recognition and removal: New twists and grooves. Mutat Res. 2005;577:55–76. doi: 10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 23.Hitomi K, Iwai S, Tainer JA. The intricate structural chemistry of base excision repair machinery: Implications for DNA damage recognition, removal and repair. DNA Repair. 2007 doi: 10.1016/j.dnarep.2006.10.004. in press. [DOI] [PubMed] [Google Scholar]
- 24.Bruner SD, Norman DP, Verdine GL. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 25.Banerjee A, Yang W, Karplus M, Verdine GL. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature. 2005;434:612–618. doi: 10.1038/nature03458. [DOI] [PubMed] [Google Scholar]
- 26.Radom CT, Banerjee A, Verdine GL. Structural characterization of human 8-oxoguanine DNA glycosylase variants bearing active site mutations. J Biol Chem. 2006 doi: 10.1074/jbc.M608989200. In press, Nov. 16 online. [DOI] [PubMed] [Google Scholar]
- 27.Banerjee A, Verdine GL. A nucleobase lesion remodels the interaction of its normal neighbor in a DNA glycosylase complex. Proc Natl Acad Sci USA. 2006;103:15020–15025. doi: 10.1073/pnas.0603644103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banerjee A, Santos WL, Verdine GL. Structure of a DNA Glycosylase Searching for DNA Lesions. Science. 2006;311:1153–1157. doi: 10.1126/science.1120288. [DOI] [PubMed] [Google Scholar]
- 29.Fromme JC, Verdine GL. DNA lesion recognition by the bacterial repair enzyme MutM. J Biol Chem. 2003;278:51543–51548. doi: 10.1074/jbc.M307768200. [DOI] [PubMed] [Google Scholar]
- 30.Blainey PC, van Oijen AM, Barnerjee A, Verdine GL, Xie XS. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc Natl Acad Sci USA. 2006;103:5752–5757. doi: 10.1073/pnas.0509723103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang YL, et al. Recognition of an Unnatural Difluorophenyl Nucleotide by Uracil DNA Glycosylase. Biochemistry. 2004;43:15429–15438. doi: 10.1021/bi0483864. [DOI] [PubMed] [Google Scholar]
- 32.Noll DM, Gogos A, Granek JA, Clarke ND. The C-terminal Domain of the Adenine-DNA Glycosylase MutY Confers Specificity of 8-Oxoguanine-Adenine Mispairs and May have evolved from MutT, an 8-oxo-dGTPase. Biochemistry. 1999;38:6374–6379. doi: 10.1021/bi990335x. [DOI] [PubMed] [Google Scholar]
- 33.Chmiel NH, Golinelli MP, Francis AW, David SS. Efficient Recognition of substrates and substrate analogs by the adenine glycosylase MutY requires the C-terminal domain. Nucleic Acids Res. 2001;29:553–564. doi: 10.1093/nar/29.2.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fromme JC, Banerjee A, Huang SJ, Verdine GL. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004;427:652–656. doi: 10.1038/nature02306. [DOI] [PubMed] [Google Scholar]
- 35.Wiederholdt CJ, Delaney MO, Pope MA, David SS, Greenberg MM. Repair of DNA Containing FapydG and its C-Nucleoside Analogue by Formamidopyrimidine DNA Glycosylase and MutY. Biochemistry. 2003;42:9755–9760. doi: 10.1021/bi034844h. [DOI] [PubMed] [Google Scholar]
- 36.Au KG, Cabrera M, Miller JH, Modrich P. Escherichia coli mutY gene product is required for specific AG to CG mismatch correction. Proc Natl Acad Sci USA. 1988;85:9163–9166. doi: 10.1073/pnas.85.23.9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Au KG, Clark S, Miller JH, Modrich P. Escherichia coli mutY gene encodes an adenine glycosylase active on G-A mispairs. Proc Natl Acad Sci USA. 1989;86:8877–8881. doi: 10.1073/pnas.86.22.8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu AL, Chang DY. A Novel Nucleotide Excision Repair for the Conversion of an A/G Mismatch to C/G Base Pair in E. coli Cell. 1988;54:805–812. doi: 10.1016/s0092-8674(88)91109-9. [DOI] [PubMed] [Google Scholar]
- 39.Radicella JP, Clark EA, Fox MS. Some mismatch repair activities in Escherichia coli. Proc Natl Acad Sci USA. 1988;85:9674–9678. doi: 10.1073/pnas.85.24.9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernards AS, Miller JK, Bao KK, Wong I. Flipping Duplex DNA Inside Out: A double base-flipping reaction mechanism by Escherichia coli MutY Adenine Glycosylase. J Biol Chem. 2002;277:20960–20964. doi: 10.1074/jbc.C200181200. [DOI] [PubMed] [Google Scholar]
- 41.Al-Tassan N, et al. Inherited variants of MYH associated with somatic G:C to T:A mutations in colorectal tumors. Nature Gen. 2002;30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 42.Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Human Molec Genet. 2001;10:721–733. doi: 10.1093/hmg/10.7.721. [DOI] [PubMed] [Google Scholar]
- 43.Sampson JR, Jones S, Dolwani S, Cheadle JP. MutYH (MYH) and colorectal cancer. Biochem Soc Trans. 2005;33:679–683. doi: 10.1042/BST0330679. [DOI] [PubMed] [Google Scholar]
- 44.Chmiel NH, Livingston AL, David SS. Insight into the Functional Consequences of Inherited Variants of the hMYH Adenine Glycosylase Associated with Colorectal Cancer: Complementation Assays with hMYH Variants and Pre-steady-state kinetics of the Corresponding Mutated E. coli. Enzymes J Mol Biol. 2003;327:431–443. doi: 10.1016/s0022-2836(03)00124-4. [DOI] [PubMed] [Google Scholar]
- 45.Livingston AL, Kundu S, Henderson-Pozzi M, Anderson DW, David SS. Insight into the Roles of Tyrosine 82 and Glycine 253 in the Escherichia coli Adenine Glycosylase MutY. Biochemistry. 2005;44:14179–14190. doi: 10.1021/bi050976u. [DOI] [PubMed] [Google Scholar]
- 46.Livingston AL. PhD in Chemistry. University of Utah; 2006. [Google Scholar]
- 47.Pope MA, Chmiel NH, David SS. Insight into the Functional Consequences of hMYH variants associated with colorectal cancer: distinct differences in the adenine glycosylase activity and the response to AP endonuclease of Y150C and G365D murine MYH. DNA Repair. 2005;4:315–325. doi: 10.1016/j.dnarep.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 48.Tominaga Y, et al. MUTYH prevents OGG1 or APEX1 from inappropriately processing its substrate or reaction product with its C-terminal domain. Nucleic Acids Res. 2004;32:3198–3211. doi: 10.1093/nar/gkh642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wooden SH, Bassett HM, Wood TG, McCullough AK. Identification of critical residues required for the mutation avoidance function of human MutY (hMYH) and implications in colorectal cancer. Cancer Lett. 2004;205:89–95. doi: 10.1016/j.canlet.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 50.Hirano S, et al. Mutator Phenotype of MutYH-null Mouse Embryonic Stem Cells. J Biol Chem. 2003;278:38121–38124. doi: 10.1074/jbc.C300316200. [DOI] [PubMed] [Google Scholar]
- 51.Lipton L, et al. Carcinogenesis in MYH-Associated Polyposis Follows a Distinct Genetic Pathway. Cancer Res. 2003;63:7595–7599. [PubMed] [Google Scholar]
- 52.Xie Y, et al. Deficiences in Mouse Myh and Ogg1 results in Tumor Predisposition and G to T Mutations in Codon 12 of the K-Ras Oncogene in Lung Tumors. Cancer Res. 2004;64:3096–3102. doi: 10.1158/0008-5472.can-03-3834. [DOI] [PubMed] [Google Scholar]
- 53.Russo MT, et al. Accumulation of the Oxidative Base Lesion 8-Hydroxyguanine in DNA of Tumor-Prone Mice Defective in Both the Myh and Ogg1 DNA Glycosylase. Cancer Res. 2004;64:4411–4414. doi: 10.1158/0008-5472.CAN-04-0355. [DOI] [PubMed] [Google Scholar]
- 54.Sieber OM, et al. Myh Deficiency Enhances Intestinal Tumorigenesis in Multiple Intestinal Neoplasia (ApcMin/+) Mice. Cancer Res. 2004;64:8876–8881. doi: 10.1158/0008-5472.CAN-04-2958. [DOI] [PubMed] [Google Scholar]
- 55.Sampson JR, et al. MYH Polyposis: a new autosomal recessive form of familial adenomatous polyposis demanding reappraisal of genetic risk and family management. Lancet. 2003;362:39–41. [Google Scholar]
- 56.Chow E, Thirlwell C, Macrae F, Lipton L. Colorectal Cancer and Inherited Mutations in Base-Excision Repair. Lancet Oncol. 2004;5:600–606. doi: 10.1016/S1470-2045(04)01595-5. [DOI] [PubMed] [Google Scholar]
- 57.Lipton L, Tomlinson I. The Multiple Colorectal Adenoma Phenotype and MYH, a excision repair gene. Clin Gastroenterol Hepatol. 2004;2:633–638. doi: 10.1016/s1542-3565(04)00286-1. [DOI] [PubMed] [Google Scholar]
- 58.Tenesa A, et al. Association of MutYH and colorectal cancer. British Journal of Cancer. 2006;95:239–242. doi: 10.1038/sj.bjc.6603239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lipton L, Tomlinson I. The genetics of FAP and FAP-like syndromes. Familial Cancer. 2006;5:221–226. doi: 10.1007/s10689-005-5673-3. [DOI] [PubMed] [Google Scholar]
- 60.Farrington SM, et al. Germline Susceptibility to Colorectal Cancer Due to Base-Excision Repair Gene Defects. Am J Hum Genet. 2005;77:112–119. doi: 10.1086/431213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fleischmann C, et al. Comprehensive Analysis of the Contribution of Germline MYH Variation of Early-Onset Colorectal Cancer. Int J Cancer. 2004;109:554–558. doi: 10.1002/ijc.20020. [DOI] [PubMed] [Google Scholar]
- 62.Strate LL, Syngal S. Hereditary colorectal cancer syndromes. Cancer Causes and Control. 2005;16:201–213. doi: 10.1007/s10552-004-3488-4. [DOI] [PubMed] [Google Scholar]
- 63.Jo WS, Chung DC. Genetics of Hereditary Colorectal Cancer. Seminars in Oncology. 2005;32:11–23. doi: 10.1053/j.seminoncol.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 64.Bodmer WF. Cancer genetics: colorectal cancer as a model. J Hum Genet. 2006;51:391–396. doi: 10.1007/s10038-006-0373-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kinzler KW, Vogelstein B. Lessons from Hereditary Colorectal Cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 66.Soreide K, Janssen EAM, Soiland H, Korner H, Baak JPA. Microsatellite instability in colorectal cancer. British Journal of Surgery. 2006;93:395–406. doi: 10.1002/bjs.5328. [DOI] [PubMed] [Google Scholar]
- 67.Lindor NM, et al. Recommendations for the Care of Individuals with an Inherited Predisposition to Lynch Syndrome. JAMA. 2006;296:1507–1517. doi: 10.1001/jama.296.12.1507. [DOI] [PubMed] [Google Scholar]
- 68.Venesio T, et al. High Frequency of MYH Gene Mutations in a Subset of Patients with Familial Adenomatous Polyposis. Gastroenterology. 2004;126:1681–1685. doi: 10.1053/j.gastro.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 69.Leite JS, et al. Is prophylactic colectomy indicated in patients with MYH-associated polyposis? Colorectal Disease. 2005;7:327–331. doi: 10.1111/j.1463-1318.2005.00811.x. [DOI] [PubMed] [Google Scholar]
- 70.Cheadle JP, Sampson JR. MUTYH-associated polyposis-From defect in base excision repair to clinical genetic testing. DNA Repair. 2006 doi: 10.1016/j.dnarep.2006.11.001. in press. [DOI] [PubMed] [Google Scholar]
- 71.Gismondi V, et al. Prevalence of the Y165C, G382D and 1395delGGA germline mutations of the MYH gene in Italian patients with adenomatous polyposis coli and colorectal adenomas. Int J Cancer. 2004;109:680–684. doi: 10.1002/ijc.20054. [DOI] [PubMed] [Google Scholar]
- 72.Isidro G, et al. Germline MutYH (MYH) Mutations in Portuguese Individuals with Multiple Colorectal Adenomas. Hum Mutat. 2004;24:353–354. doi: 10.1002/humu.9282. [DOI] [PubMed] [Google Scholar]
- 73.Bai H, et al. Functional characterization of two human MutY homolog (hMYH) missense mutations (R227W and V232F) that lie within the putative hMSH6 binding domain and are associated with hMYH polyposis. Nucleic Acids Res. 2005;33:597–604. doi: 10.1093/nar/gki209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bai H, et al. Functional characterization of human MutY homolog (hMYH) missense mutation (R231L) that is linkied with hMYH-associated polyposis. Cancer Lett. 2006 doi: 10.1016/j.canlet.2006.09.016. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alhopuro P, et al. A novel functionally deficient MYH variant in individuals with colorectal adenomatous polyposis. Human Mutat. 2005;26:393. doi: 10.1002/humu.9368. [DOI] [PubMed] [Google Scholar]
- 76.Klunglund A, et al. Accumulation of premutagenic DNA lesions in mice defective in removal of oxidative base damage. Proc Natl Acad Sci USA. 1999;96:13300–13305. doi: 10.1073/pnas.96.23.13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Osterod M, et al. A global DNA repair mechanism involving Cockayne syndrome B (CSB) gene product can prevent the in vivo accumulation of endogenous oxidative DNA base damage. Oncogene. 2002;21:8232–9239. doi: 10.1038/sj.onc.1206027. [DOI] [PubMed] [Google Scholar]
- 78.Osterod M, et al. Age-related and tissue-specific accumulation of oxidative DNA base damage in 7,8-dihydro-8-oxoguanine-DNA glycosylase (Ogg1) deficient mice. Carcinogenesis. 2001;22:1459–1463. doi: 10.1093/carcin/22.9.1459. [DOI] [PubMed] [Google Scholar]
- 79.Sunesen M, Stevnsner T, Brosh RM, Dianov GL, Bohr VA. Global genome repair of 8-oxoG in hamster cells requires a functional CSB gene product. Oncogene. 2002;21:3571–3578. doi: 10.1038/sj.onc.1205443. [DOI] [PubMed] [Google Scholar]
- 80.Cadet J, Decarroz C, Wang SY, Midden WR. Mechanisms and products of photosensitized degradation of nucleic acids and related model compounds. Isr J Chem. 1983;1983:420–429. [Google Scholar]
- 81.Ravanat JL, Cadet J. Reaction of Singlet Oxygen with 2′-deoxyguanosine and DNA. Isolation and characterization of the main oxidation products. Chem Res Toxicol. 1995;8:379–388. doi: 10.1021/tx00045a009. [DOI] [PubMed] [Google Scholar]
- 82.Ravanat JL, Berger M, Bernard F, Langlois R, Ouellet R. Phthalocyanine and naphthalocyanine photosensitized oxidation of 2′-deoxyguanosine: distinct type 1 and type II products. Photochem Photobiol. 1992;55:809–814. [Google Scholar]
- 83.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of Spiroiminodihydantoin as a Product of One-Electron Oxidation of 8-Oxo-7,8-dihydroguanosine. Org Lett. 2000;2:613–616. doi: 10.1021/ol9913643. [DOI] [PubMed] [Google Scholar]
- 84.Luo W, Muller JG, Rachlin EM, Burrows CJ. Characterization of Hydantoin Products from One-Electron Oxidation of 8-Oxo-7,8-dihydroguanosine in a Nucleoside Model. Chem Res Toxicol. 2001;14:927–938. doi: 10.1021/tx010072j. [DOI] [PubMed] [Google Scholar]
- 85.Luo W, Muller JG, Burrows CJ. The pH-Dependent Role of Superoxide in Riboflavin-Catalyzed Photooxidation of 8-Oxo-7,8-dihydroguanosine. Org Lett. 2001;3:2801–2804. doi: 10.1021/ol0161763. [DOI] [PubMed] [Google Scholar]
- 86.Burrows CJ, et al. Structure and potential mutagenicity of new hydantoin products from guanosine and 8-oxo-7,8-dihydroguanosine oxidation by transition metals. Environ Health Perspect. 2002;110:713–717. doi: 10.1289/ehp.02110s5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Niles JC, Wishnok JS, Tannenbaum SR. Spiroiminodihydantoin is the Major Product of the 8-Oxo-7,8-dihydroguanosine Reaction with Peroxynitrite in the Presence of Thiols and Guanosine Photooxidation by Methylene Blue. Org Lett. 2001;3:963–6. [PubMed] [Google Scholar]
- 88.Adam W, Arnold MA, Grune M, Nau WM, Pischel U, Saha-Moller CR. Spiroiminodihydantoin is a Major Product in the Photooxidation of 2′-Deoxyguanosine by the Triplet States and Oxyl Radicals Generated from Hydroxyacetophenone Photolysis and Dioxetane Thermolysis. Org Lett. 2002;4:537–540. doi: 10.1021/ol017138m. [DOI] [PubMed] [Google Scholar]
- 89.Leipold MD, Muller JG, Burrows CJ, David SS. Removal of Hydantoin Products of 8-Oxoguanine Oxidation by the Escherichia coli DNA Repair Enzyme, Fpg. Biochemistry. 2000;39:14984–14992. doi: 10.1021/bi0017982. [DOI] [PubMed] [Google Scholar]
- 90.Kornyushyna O, Berges AM, Muller JG, Burrows CJ. In vitro nucleotide misinsertion opposite the oxidized guanosine lesions spiroiminodihydantoin and guanidinohydantoin and DNA synthesis past the lesions using Escherichia coli DNA polymerase I (Klenow fragment) Biochemistry. 2002;41:15304–15314. doi: 10.1021/bi0264925. [DOI] [PubMed] [Google Scholar]
- 91.Kornyushyna O, Burrows CJ. Effect of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin on proofreading by Escherichia coli DNA polymerase I (Klenow Fragment) in different sequence contexts. Biochemistry. 2003;42:13008–13018. doi: 10.1021/bi0350755. [DOI] [PubMed] [Google Scholar]
- 92.Henderson PT, et al. The hydantoin lesions from oxidation of 7,8-dihydro-8-oxoguanine are potent sources of replication errors in vivo. Biochemistry. 2003;42:9257–9262. doi: 10.1021/bi0347252. [DOI] [PubMed] [Google Scholar]
- 93.Leipold MD, Workman H, Muller JG, Burrows CJ, David SS. Recognition and Removal of Oxidized Guanines in Duplex DNA by the Base Excision Repair Enzymes hOGG1, yOGG1 and yOGG2. Biochemistry. 2003;42:11373–11381. doi: 10.1021/bi034951b. [DOI] [PubMed] [Google Scholar]
- 94.Hazra TK, et al. Repair of hydantoins, one electron oxidation product of 8-oxoguanine, by DNA glycosylases of Escherichia coli. Nucleic Acids Res. 2001;29:1967–1974. doi: 10.1093/nar/29.9.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wallace SS, Bandaru V, Kathe SD, Bond JP. The enigma of endonuclease VIII. DNA Repair. 2003;2:441–453. doi: 10.1016/s1568-7864(02)00182-9. [DOI] [PubMed] [Google Scholar]
- 96.Hailer MK, Slade PG, Martin BD, Sugden KD. Nei deficient Escherichia coli are sensitive to chromate and accumulate the oxidized guanine lesion spiroiminodihydantoin. Chem Res Toxicol. 2005;18:1378–1383. doi: 10.1021/tx0501379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bandaru V, Sunkara S, Wallace SS, Bond JP. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair. 2002;1:517–529. doi: 10.1016/s1568-7864(02)00036-8. [DOI] [PubMed] [Google Scholar]
- 98.Hazra TK, et al. Identification and Characterization of a human DNA glycosylase for repair of modified oxidatively damaged DNA. Proc Natl Acad Sci USA. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hazra TK, et al. Identification of a Novel Human DNA Glycosylase for Repair of Cytosine-derived Lesions. J Biol Chem. 2002;277:30417–30420. doi: 10.1074/jbc.C200355200. [DOI] [PubMed] [Google Scholar]
- 100.Morland I, et al. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002;30:4926–4936. doi: 10.1093/nar/gkf618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Doublie S, Bandaru V, Bond JP, Wallace SS. The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proc Natl Acad Sci USA. 2004;101:10284–10289. doi: 10.1073/pnas.0402051101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Katafuchi A, et al. Differential Specificity of Human and Escherichia coli Endonuclease III and VIII Homologues for Oxidative Base Lesions. J Biol Chem. 2004;279:14464–14471. doi: 10.1074/jbc.M400393200. [DOI] [PubMed] [Google Scholar]
- 103.Dou H, Mitra S, Hazra TK. Repair of Oxidized Bases in DNA Bubble Structures by Human DNA Glycosylases NEIL1 and NEIL2. J Biol Chem. 2003;278:49679–49684. doi: 10.1074/jbc.M308658200. [DOI] [PubMed] [Google Scholar]
- 104.Hailer KM, Slade PG, Martin BD, Rosenquist TA, Sugden KD. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the base excision repair glycosylases NEIL1 and NEIL2. DNA Repair. 2005;4:41–50. doi: 10.1016/j.dnarep.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 105.Krishnamurthy N, Zhao X, Muller JG, Burrows CJ, David SS. unpublished results. [Google Scholar]
- 106.Das A, Hazra TK, Boldogh I, Mitra S, Bhakat KK. Induction of the Human Oxidized Base-specific DNA Glycosylase NEIL1 by Reactive Oxygen Species. J Biol Chem. 2005;280:35272–35280. doi: 10.1074/jbc.M505526200. [DOI] [PubMed] [Google Scholar]
- 107.Rosequist TA, et al. The novel DNA glycosylase, NEIL1, protects mammalian cells from radiation-mediated cell death. DNA Repair. 2003;2:581–591. doi: 10.1016/s1568-7864(03)00025-9. [DOI] [PubMed] [Google Scholar]
- 108.Shinmura K, et al. Inactivating mutations of the human base excision repair gene NEIL1 in gastric cancer. Carcinogenesis. 2004:2311–2317. doi: 10.1093/carcin/bgh267. [DOI] [PubMed] [Google Scholar]
- 109.Vartanian V, et al. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc Natl Acad Sci USA. 2006;103:1864–1869. doi: 10.1073/pnas.0507444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Guan Y, Manuel RC, Arvai AS, Parikh SS, Mol CD, Miller JH, Lloyd RS, Tainer JA. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nature Struct Biol. 1998;5:1058–1064. doi: 10.1038/4168. [DOI] [PubMed] [Google Scholar]
- 111.Lukianova OL, David SS. A role for iron-sulfur clusters in DNA repair. Curr Opin Chem Biol. 2005;9:145–151. doi: 10.1016/j.cbpa.2005.02.006. [DOI] [PubMed] [Google Scholar]