

Abstract
Purpose
Aberrant nuclear activation and phosphorylation of the canonical NF-κB subunit RELA/p65 at Serine-536 by Inhibitor Kappa B Kinase is prevalent in head and neck squamous cell carcinoma (HNSCC), but the role of other kinases in NF-κB activation has not been well defined. Here, we investigated the prevalence and function of p65-Ser276 phosphorylation by Protein Kinase A (PKA) in the malignant phenotype, gene transactivation, and as a potential target for therapy.
Experimental Design
Phospho and total p65 protein expression and localization was determined in HNSCC tissue array and in cell lines. The effects of PKA inhibitor H-89 on cell proliferation and cell cycle and of H-89 and PKA specific siRNA knockdown on NF-κB activation and downstream gene expression were examined.
Results
Nuclear NF-κB p65 phosphorylated at Ser276 was prevalent in HNSCC and adjacent dysplastic mucosa, but localized to the cytoplasm in normal mucosa. In HNSCC lines, TNF-α significantly increased while H-89 inhibited constitutive and TNF-α induced nuclear p65-Ser276 phosphorylation, and significantly suppressed NF-κB and target gene IL-8 reporter activity. Knock down of PKA by siRNA inhibited NF-κB, IL-8 and BCL-XL reporter gene activities. H-89 suppressed cell proliferation, induced cell death and blocked the cell cycle in G1/S phase. Consistent with its biological effects, H-89 down-modulated expression of NF-κB related genes Cyclin D1, BCL2, BCL-XL, COX2, IL-8, and VEGF, as well as induced cell cycle inhibitor p21CIP1/WAF1, while suppressing proliferative marker Ki67.
Conclusions
NF-κB RELA Ser276 phosphorylation by PKA promotes the malignant phenotype and holds potential as a therapeutic target in HNSCC.
Keywords: NF-κB p65 Serine276, phosphorylation, PKA, H-89, head and neck cancer
Introduction
Nuclear factor kappa B (NF-κB) is a signal transcription factor that has emerged as an important modulator of altered gene expression programs and malignant phenotype in development and progression of cancer. We have reported previously that NF-κB is aberrantly activated and promotes altered expression of multiple genes important in the malignant phenotype of human and murine HNSCC (1, 2). The canonical mechanism for NF-κB nuclear translocation and activation involves the phosphorylation and subsequent degradation of its cytoplasmic inhibitor kappa B (IκB) by a trimeric Inhibitor-kappa B Kinase (IKK) α/β/γ complex in response to wide range of stimuli such as oncogenic viruses, carcinogens, growth factors and cytokines. Degradation of IκB results in the liberation of the p65-p50 heterodimer for its translocation to the nucleus where it transactivates target genes (3). The classical pathway of activation of NF-κB by physical dissociation of p65-p50 subunits from IκBα and subsequent nuclear translocation has been well studied (4, 5), but the differential dynamics of NF-κB transactivation following the phosphorylation of p65 at various sites is increasingly being appreciated. There is evidence that IKK and various other kinases induce phosphorylation of p65 at Ser 536, a post translational modification critical for the transactivation of NF-κB in response to various stimuli (6–12). Likewise, another site, p65-Ser276 has been shown to be phosphorylated by the catalytic subunit of protein kinase A (PKA) (13, 14) and Mitogen and Stress Activated Protein Kinase-1 (MSK-1) (15, 16). The precise role of phosphorylation at Ser 276 in NF-κB gene transactivation and the malignant phenotype in HNSCC is yet unclear.
The PKA families of proteins are cyclic adenosine monophosphate(c-AMP) dependent holoenzymes found in mammalian cells. The PKA holoenzyme consists of a heterotetramer of two homodimers of the regulatory (R) subunits (RIα or RIβ and RIIα or RIIβ) and two catalytic subunits (Cα, Cβ or Cγ), resulting in the formation of two isozymes, type I (RI) and type II (RII). Binding of cAMP to the regulatory subunits releases the catalytic subunits, which act as serine threonine kinases that phosphorylate target molecules to control a variety of cellular functions (17). Typically, ligand binding to G-Protein-Coupled Receptors (GPCRs) activates a stimulatory G-protein. The activated G-protein interacts with adenylate cyclase causing accumulation of cyclic AMP (cAMP). Active PKA catalytic subunits are released following cAMP binding to the regulatory subunits. The active catalytic subunits phosphorylate targets in both the cytoplasm and the nucleus. The active catalytic subunits phosphorylate targets in both the cytoplasm and the nucleus. H-89, N-[2-(bromocinnamylamino) ethyl]-5-isoquinolinesufonamide, is a potent synthetic inhibitor of PKA and has been used extensively for the evaluation of the role of PKA in various tissue types (18).
Enhanced expression of PKA has been associated with malignant transformation and cell proliferation in cell lines from various organ systems (19). PKA has been shown to stimulate the transcriptional activity of NF-κB by phosphorylation of the p65 subunit on Ser 276 (13, 14). TNF-α, a potent stimulator of NF-κB in many cell types including HNSCC, has been shown to induce phosphorylation of p65 at Ser 276 in a reactive oxygen species (ROS) dependent manner (20). Of note, several GPCRs have also recently been identified and shown to contribute to the malignant phenotype in HNSCC, including over-expression of H and K-RAS (21–24)
Here, we have examined the prevalence of phosphorylation of p65 at the Ser 276 site in HNSCC tissue array and the contribution of PKA in site-specific phosphorylation, cellular distribution and transactivation of NF-κB. We have also studied the modulation of NF-κB dependent genes implicated in the pathogenesis of HNSCC, and the functional effects of inhibition on cell proliferation and cell cycle.
Material and Methods
Cell Culture
HNSCC cell lines from the University of Michigan squamous cell carcinoma (UMSCC) series were obtained from Dr. T.E. Carey (University of Michigan, Ann Arbor, MI). UMSCC-6 was derived from biopsy of a primary carcinoma of the tongue and 11A from a primary T2N2aM0 hypopharyngeal carcinoma. Cells were maintained in Eagle’s Minimal Essential Media (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum, penicillin, streptomycin and L-glutamine (complete media) under standard growth conditions (37°C, 5% CO2, humidified atmosphere).
Immunohistochemistry (IHC) and scoring
Anonymous archived samples of HNSCC were obtained from the Cancer Tissue Network. Tissue arrays from de-identified donors were obtained from Cybrdi, Inc. (Gaithersburg, MD), and Dr. Alfredo Molinolo and J. Silvio Gutkind, National Institute of Dental and Craniofacial Research, NIH, and studied under an exemption from IRB review by the Office of Human Subjects Research, NIH. The Cybridi array included 20 HNSCC in triplicate and 6 normal mucosa in duplicate. The NIDCR array contained 197 evaluable HNSCC tumor sections from subjects worldwide, including the U.S., Thailand, Mexico, Argentina, China, India, Japan and South Africa. Detailed description of the arrays, H&E staining, immunohistochemistry, and scoring are described in Supplemental Methods.
Western blot
Whole cell, nuclear, and cytoplasmic lysates were obtained using an extraction kit from Active Motif (Carlsbad, CA) and analyzed as described in Supplemental Methods. Briefly, rabbit polyclonal anti phospho p65(Ser 276) 1:500 (Cat #3037), polyclonal Phospho-p65(Ser536) 1:500 (Cat #3031), and polyclonal PKACα1:1000 (Cat #4782) were obtained from Cell Signaling Technology (Danvers, MA). NF-κB p65 (sc-109) was obtained from Santa Cruz and used at 1:500 dilution. Goat anti-rabbit secondary was used at 1:2000 dilution (Chemicon, Cat # AP132P).
Reporter gene assay
Luciferase reporter plasmids used included a 5X NF-κB promoter binding consensus sequence (Stratagene, La Jolla, CA), IL-8 promoter (−133 bp, ref. 25) and the BCL-XL promoter sequence (−1,498 to + 427), which was PCR amplified, sequence confirmed and constructed with luciferase reporter gene plasmid using pBV-Luc by Dr. Lin Zhang (University of Pittsburgh, PA). Details of the experiment are as described in Supplementary Methods.
MTT cell proliferation assay
Cells were plated in quadruplicate onto a 96-well plate at 3 × 103 cells per well in 100μL of complete MEM. The next day, cells were exposed to H-89, and cell proliferation was measured every subsequent day using a MTT Cell Proliferation kit (Roche Diagnostics). The absorbance was measured by a microplate reader at a wavelength of 570 nm.
Flow Cytometry
Cells treated with 20 μM H-89 were incubated with the drug for 12 hours and both monolayer and nonadhesive cells were collected and counted by hemocytometer with trypan blue solution (Invitrogen). The cells were stained with propidium iodide provided by Cycletest Plus DNA Reagent kit (Becton Dickinson, San Jose, CA) following the manufacturer’s suggestions. DNA staining was quantified by FACScan flow cytometer (Becton Dickinson) using CellQuest software (Becton Dickinson). Cell cycle and DNA degradation of cells were analyzed by Diva Rsoftware built into the flow cytometer.
PKAc small interfering RNA
Cells were grown to 60% confluence and transfected using 30 nm of pooled siRNA TriFECTa DsiRNA duplex kit (Integrated DNA Technologies, Coralville, IA) using Lipofectamine 2000 (Invitrogen) at 1:50 concentration for 72 hours. Knockdown efficiency was assessed by RT-PCR with PKA specific primer probes (Applied Biosystems) and by Western blot with anti-PKAc specific antibodies.
Real time RT-PCR
Total RNA was isolated with Trizol (Invitrogen) and purified with the RNeasy Midi kit (Qiagen Inc., Valencia, CA) according to manufacturer’s instructions. Total RNA was converted to cDNA using the High-Capacity cDNA Archive kit according to manufacturer’s instructions (Applied Biosystems, Foster City, CA). RT-PCR for gene expression was achieved with Assays-on-Demand™ Gene Expression Assay from Applied Biosystems. (for details see Supplemental Methods)
Results
NF-κB p-p65 (Ser 276) localizes to the nucleus of HNSCC and adjacent dysplastic epithelium, but is restricted to the cytoplasm of normal mucosa
In the process of testing several specific antibodies for IHC detection of NF-κB in HNSCC, we detected staining and differential distribution for p-p65 (Ser 276) in an HNSCC of the lip and adjacent skin and mucosa (Fig. 1A). p-p65 (Ser 276) was found to be localized to the nucleus in tumor tissue (Fig. 1A–a) and in adjacent dysplastic epithelium (Fig. 1A–b) with little or no presence in the cytoplasm, whereas in surrounding normal mucosa, it was selectively localized to the cytoplasm. (Fig. 1A–c). In order to determine the prevalence, intensity, and distribution of p65 phosphorylated at Ser 276 in HNSCC, a tissue array containing 20 HNSCC and 6 normal mucosa controls were immunostained (Fig. 1B). The difference in staining intensities, measured as a histoscore (see Supplemental Methods) for the nuclei and cytoplasm, was compared between tumor and normal tissue using a two-sided Wilcoxon rank sum test. Significant differences were noted between normal and tumor samples in the nucleus (p= 0.02) and in the cytoplasm (p<0.001) (Fig. 1C, left and middle panels). Though the localization of staining was predominantly nuclear in tumor tissues, there was demonstrable heterogeneity with respect to the intensity (Fig. 1B). 75% of sections (15/20) showed some degree of nuclear staining and no detectable staining was found in 25% (5/20) (Fig 1C, right panel). Cytoplasmic but not nuclear staining was found in all six normal mucosa (Fig 1C, middle panel).
Figure 1. Phospho p65 (Ser 276) is localized to the nucleus in HNSCC and peritumoral dysplastic epithelium but is retained in the cytoplasm in normal mucosa.
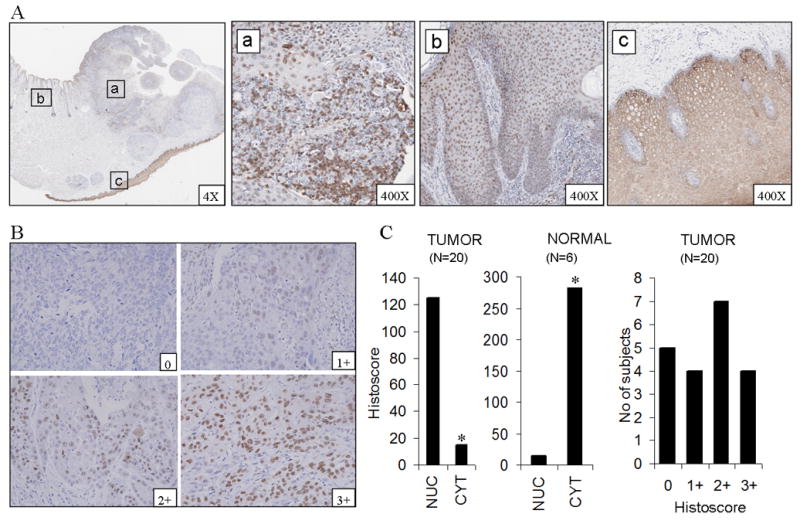
(A) Immunostaining for p-p65 (Ser 276) from different areas of the same section of carcinoma of the lip shows nuclear localization in tumor tissue (a) and adjacent dysplastic epithelium (b). Normal mucosa in the same section from a site distant to the tumor shows exclusively cytoplasmic localization (c). (B) shows different intensities of nuclear staining in tumor tissue. (C) Graphs show staining intensity, measured as histoscore, in the nuclear and cytoplasmic compartments in tumors and normal mucosae on the tissue array (left and middle panels). A variable pattern of staining intensity is demonstrated in tumors with essentially nuclear localization (right panel). Magnification 400X. *significant difference was noted between normal (n=6) and tumors (n=20) samples in the nucleus (p=0.02) and in the cytoplasm (p<0.001) by Wilcoxon rank sum test.
To further establish if the findings presented with the 20 tumor tissue array used are of wider prevalence and potential as a biomarker, we have analyzed the level of expression of p65(Ser276) on a more extensive head and neck cancer tissue array. This array contained 197 evaluable HNSCC tumor sections from subjects worldwide (see Supplemental Methods). The variability and prevalence of nuclear staining for p-p65(Ser 276) staining in this larger array was similar the that observed in our initial series. Of 197 samples, 74.6 % of samples showed some degree of nuclear staining (Supplemental Figure 1). Overall, 39.6% showed low (histoscore 1-100), 27.4% showed moderate (histoscore 101-200) and 7.6 % showed high (histoscore 201-300) and 25.4% did not show any expression of p-p65 (Ser276) in the nuclei of tumors. Thus, a pattern of nuclear staining was prevalent in HNSCC and adjacent dysplastic mucosa, when compared with several non-malignant mucosal epithelial specimens.
HNSCC cell lines exhibit constitutive and TNF-α-inducible phosphorylation of p65 (Ser 276)
To further dissect the mechanisms of p65 (Ser 276) phosphorylation, we selected two HNSCC cell lines, UMSCC-6 and UMSCC-11A, previously shown to have to have increased p65 activation levels compared with normal keratinocytes (26). The constitutive and TNF-α-induced expression and distribution of phospho p65 (Ser 276) was studied. In HNSCC cell lines ex vivo, relatively weak constitutive nuclear p-p65 (Ser 276) is observed by IHC (Fig. 2A), potentially due to a lack of TNF-α expression by these cell lines (27), which has been shown to be present in the tumor microenvironment (28). Consistent with this hypothesis, treatment with recombinant TNF-α significantly enhanced nuclear p-p65 (Ser 276) staining above the constitutive level in both cell lines (Fig 2A) as early as 30 minutes of treatment.
Figure 2. HNSCC cell lines express constitutive and TNF activated phosphorylated p65 (Ser 276).
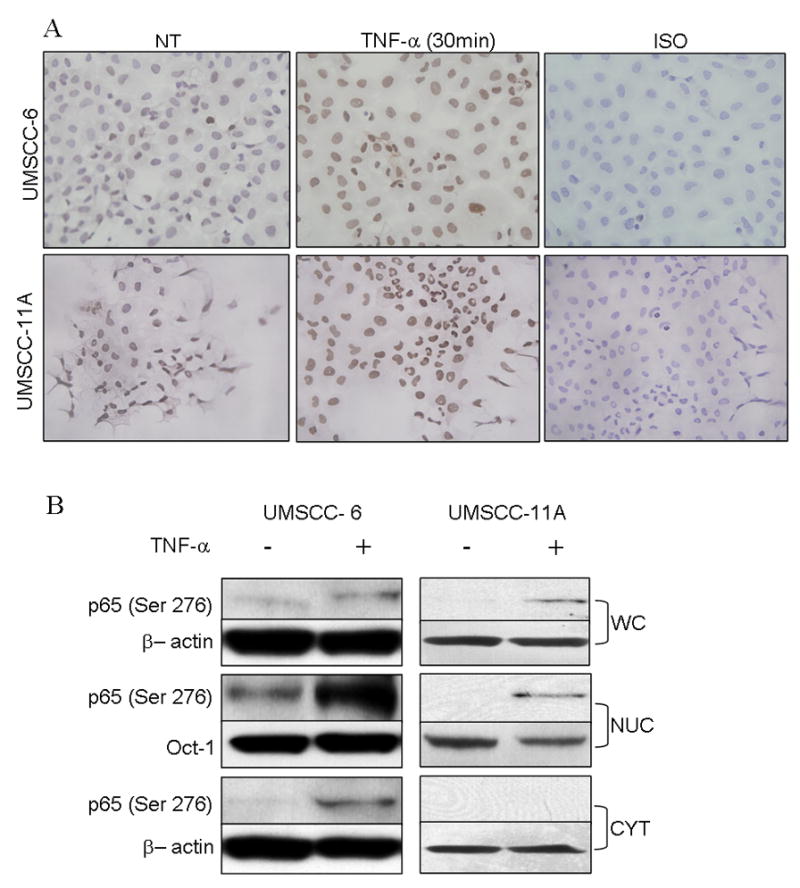
(A) UMSCC-6 (top) and UMSCC-11A cells (bottom) grown in chamber slides, fixed and immunostained for phospho p65(Ser 276). Cells show nuclear localization of p-p65 (Ser 276) which is increased by pretreating the cells with 10 ng/ml of TNF-α for 30 minutes. (B) Immunoblot of whole cell, nuclear and cytoplasmic extracts of UMSCC-6 with and without activation with 10 ng/ml TNF-α for 30 minutes.
To further analyze the relative expression and distribution of p-p65 (Ser 276) in these cell lines, Western blot analysis was performed in whole cell lysates and cell fractions (Fig 2B). In UMSCC-6, minimal, and in UMSCC-11A, undetectable levels of p-p65 (Ser 276) were observed in whole cell lysates (Fig 2B), and TNF treatment enhanced p65 (Ser 276) phosphorylation. In nuclear extracts, a higher basal and TNF-α inducible p-p65 (Ser 276) were observed in UMSCC-6 as compared to the relatively weaker expression and induction detected in UMSCC-11A. p-p65 (Ser 276) was only observed in the cytoplasmic fractions of UMSCC-6 and with TNF-α treatment. The above experiments confirm that localization of p65 (Ser 276) is predominantly nuclear and phosphoylation levels are enhanced on treatment with TNF-α.
PKA inhibitor H-89 significantly inhibits the activation of p-p65 (Ser 276) without affecting nuclear translocation of p65
We found that H-89 treatment of UMSCC-6 resulted in a dose dependent decrease in constitutive and TNF-α induced expression of p-p65 (Ser 276) in immunoblots (Fig. 3A). Furthermore, UMSCC-6 cells were treated with H-89 for varying durations between 1 and 24 hours, without or with activation with TNF-α. H-89 inhibited constitutive p-p65 (Ser 276) at 1 hour of treatment and more significantly at 6 hours, persisting for 24 hours (Fig. 3B, left panels). Similar decrease was noted in TNF-α induced samples initially followed by fluctuations at later time courses (Fig. 3B, left panels), consistent with oscillations reported for TNF-α induced cytoplasmic-nuclear shuttling due to degradation and resynthesis of IκB (29). p-p65 (Ser 536) levels showed fluctuations over time with no effective decrease till the 24 hour time point (Fig 3B, left panel). With TNF-α, p-p65 (Ser 536) levels are induced while no change in expression level is effected by H-89. Constitutive and induced total p65 levels remain unchanged with H-89 over the time course. The above results show that PKA inhibitor H-89 attenuates both constitutive and TNF activated phosphorylation of p65 at Ser 276. H-89 produced fluctuation in the basal levels of p-p65 (Ser 536), without affecting TNF-α induced levels of p- p65 (Ser 536) or total p65 in the cells.
Figure 3. PKA inhibitor H-89 blocks formation of Phospho p65 (Ser276) without impairing formation of Phospho p65(Ser 536) or affecting nuclear translocation of p65.
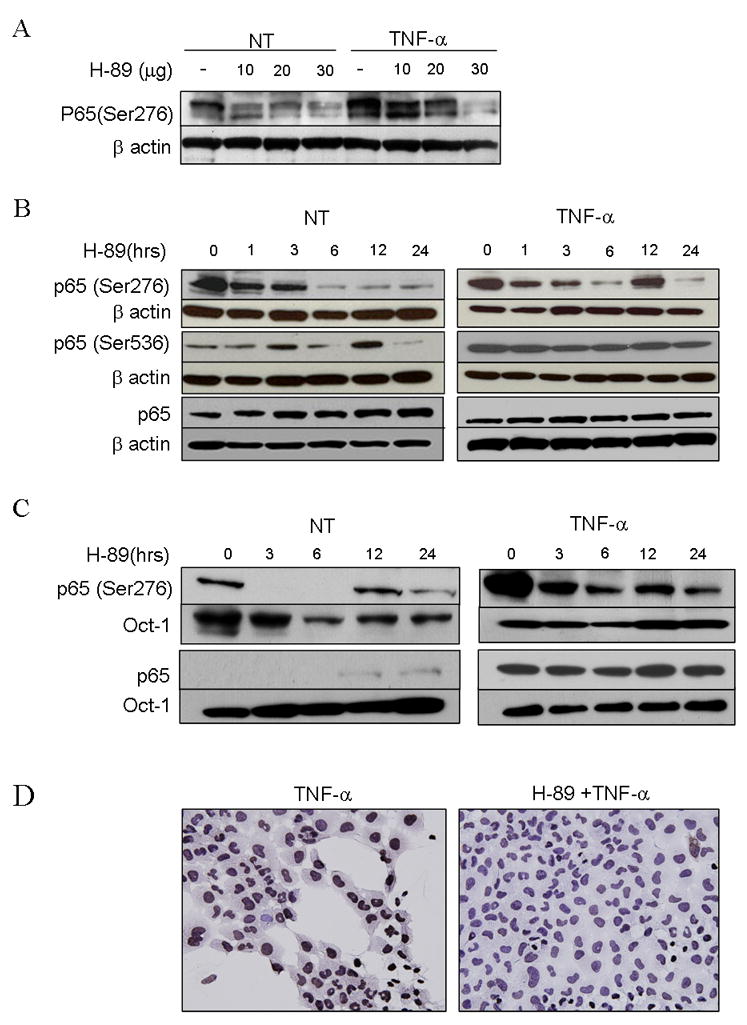
(A) western blot using 30 μg whole cell extracts of UMSCC-6 treated with 20 μM H-89 for varying times as indicated, with (right panel) and without (left panel) activation with 10ng/ml of TNF-α for 30 minutes prior to extraction of proteins, and probed with p-p65 (Ser276), p-p65 (Ser536) antibody and total p65. (B) UMSCC-6 cells were treated with varying doses of H-89 as shown for 3 hours without and with activation with TNF-α. Whole cell lysates obtained were blotted and probed with phospho p65 (Ser276) with beta actin as loading control. (C) shows nuclear extracts from UMSCC-6 cells treated similarly with H-89 with and without activation with TNF-α and probed for p65 and phospho p65(Ser 276). Oct-1 was used as loading control. (D) shows UMSCC-6 cells grown on chamber slides, activated with TNF for 30 minutes, fixed with paraformaldehyde and immunostained for p-p65(Ser 276) compared with and without pretreatment with H-89 for 1 hour. (Magnification 400×)
In order to study whether the H-89-induced time-dependent decrease in phospho-p65 (ser276) was due to decreased nuclear translocation, we examined the effect of H-89 treatment on nuclear translocation. A rapid translocation of p65 within 30 minutes of TNF treatment (Fig. 3C, time 0) was observed, which was unaffected by pretreatment with H-89 (Fig. 3C). p-p65(Ser 276) disappeared from the nucleus by 3 hours of treatment with H-89 and reappeared at 12 hours, showing oscillations in levels similar to observations in the whole cell levels. TNF-α activated p-p65 (ser276) levels also decreased within 3 hours of treatment with H-89. The results were further confirmed by immunohistochemistry, where UMSCC-6 cells treated with 20 μm H-89 and activated for 30 min with TNF-α showed reduced expression of nuclear p-p65 (Ser 276) compared with untreated cells activated with TNF-α (Fig. 3D). Evidence from the above experiments suggest that H-89 induced inhibition of PKA causes an inhibition of site specific phosphorylation at Ser 276 with less prominent effects on the phosphorylation at Ser 536, or nuclear translocation of p65 mediated by IKK.
Inhibition of PKA using H-89 or specific siRNA knockdown inhibits transactivation of NF-κB and its dependent genes
In order to study the role of inhibition of site specific phosphorylation at Ser 276 on p65 on NF-κB transactivation, we studied the effect of H-89 on reporter activity in unstimulated and TNF-α stimulated UMSCC-6 cells. TNF-α showed a highly significant increase in NF-κB reporter activity from the baseline. Treatment with H-89 for 24 hours caused a significant decrease in basal and TNF-α induced reporter activity (Fig. 4A, left panel). The effects of H-89 on NF-κB activation was further tested using the IL-8 proximal promoter containing −133 bp form the transcription start site (25). H-89 significantly inhibited reporter activity of IL-8 in both unstimulated and TNF-α stimulated cells (Fig. 4A, right panel). However, DNA binding of p65 and other NF-κB subunits p50, p52, Rel B and c-Rel to an NF-κB consensus oligonucleotide sequence was not affected in unstimulated and TNF-α stimulated nuclear extracts of UMSCC-6 treated with H-89 for 24 hours.
Figure 4. Inhibition of PKA, both by drug and siRNA transfection inhibits reporter activity of NFkB and its dependent genes.
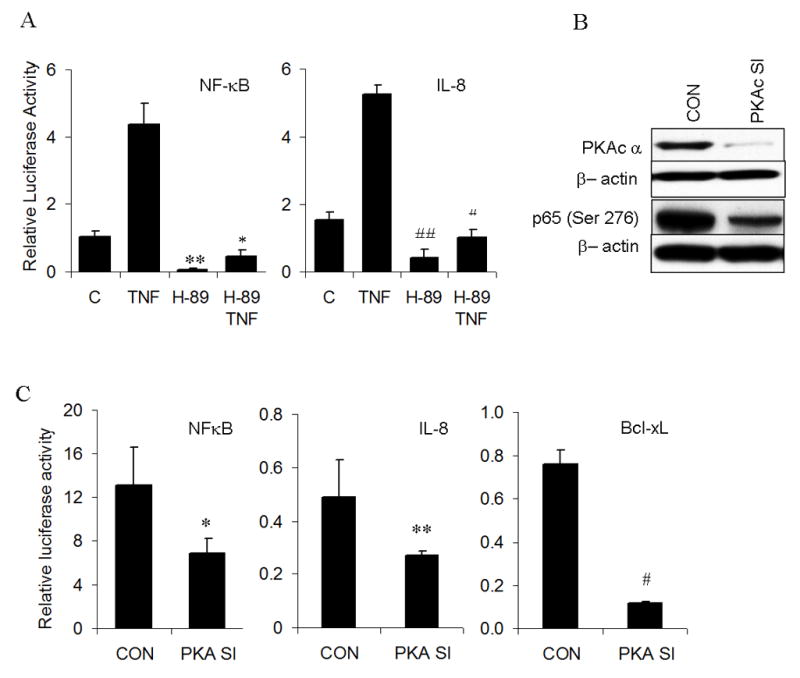
(A) UMSCC-6 cells were transfected with NF-κB consensus reporter (0.15 μg/well or IL-8 reporter at 0.2 μg/well and beta gal reporter (0.02 μg/well) following which transfection medium was replaced with antibiotic free medium containing 20μm H-89 or 20μm H-89 and 10ng/ml TNF. Cells were lysed 24 hours post transfection and reporter activity measured as relative light units. Readings shown is an average of three independent experiments (**p=10−5; * p=2.6×10−8 for NFkB. # p=1.3 × 10−5; ## p= 4.7 ×10−11 for IL-8). (B) shows immunoblots of whole cell extracts from UMSCC-6 transfected with 30nM of PKAc siRNA or control siRNA and probed for PKACα or p-p65(Ser 276). (C) Reporter activity of NF-κB, IL-8 and BCL-XL genes following co-transfection of corresponding reporters with 30nM PKAc siRNA for 72 hours. Equimolar dose of scrambled siRNA was used as control. All values represent the average value of three independent observations. (* p= 0.0002; ** p= 0.003; # p= 4.2 × 10−10)
In order to confirm the specificity of effects of H-89 on PKA, we knocked down expression of the catalytic subunit of PKA (PKAc) by siRNA transfection. Significant knockdown of PKA levels was achieved at 72 hours of transfection. There was also significant, though not complete, inhibition of phosphorylation of p65 at serine 276 (Fig. 4B). This inhibition significantly decreased NF-κB reporter activity and correspondingly suppressed reporter activity for NF-κB dependent genes IL-8 and Bcl-xL (Fig. 4C). However, siRNA mediated knockout of mitogen and stress activated kinase-1(MSK-1), failed to show a statistically significant decrease in NF-κB reporter activity (data not shown), indicating that PKA is a major contributor to transactivation of NF-κB in the UMSCC 6 cells.
PKA inhibition by H-89 modulates expression of multiple genes implicated in the development and progression of HNSCC
NF-κB has been shown to modulate a broad panel of gene expression which promotes tumor development and progression, including cell cycle dysregulation and proliferation, apoptosis, inflammation and angiogenesis (1, 2, 30). Here we show that prominent effects on gene modulation at different time points. We observed increased levels of p21CIP1/WAF1 starting 3 hours after treatment, which is a p53-regulated cyclin dependent kinase that promotes cell cycle arrest, and a decrease in known NF-κB modulated gene Cyclin D1, known to promote transition of cells through the G1 phase of cell cycle. H-89 was shown to also inhibit expression of known NF-κB-regulated prosurvival genes BCL-2 and BCL-XL, and pro-inflammatory and angiogenesis mediators such as COX-2 (31–33), IL-8 and VEGF. Consistent with this, an inhibitory effect on cell proliferation was noted by a decrease in Ki-67 mRNA levels at later time points. The above experiments demonstrate that H-89 inhibition of PKA induced phosphorylation of p65 at Ser 276 has important effects in modulating genes, to enhance cell cycle arrest and apoptosis, and inhibition of inflammation and angiogenesis.
H-89 inhibits cell proliferation in a dose dependent manner and induces cell death and G1/S phase arrest in cell cycle
We further analyzed the effects of H-89 on proliferation of HNSCC cell lines. Both UMSCC-6 and UMSCC-11A cell lines showed a dose dependent inhibition in proliferation with and IC50 of approximately 20 micromoles (Fig 6A). H-89 induced cell death was observed as early as 12 hours after treatment, as evidenced from and increase of cells in the sub G0 phase from 20.8% to 46.82 % (Fig. 6B). Further analysis of the viable cell population showed an increase in H-89 treated cells at the G0/G1 phase compared to untreated controls (71.4% and 56.7%). H-89 treatment also caused a reduction in cells at the S (12.8% and 7.7%) and G2/M phases (29.2% and 17.6%). Overall, H-89 was shown to induce cytotoxicity with cell cycle arrest at the G0/G1 phase, and decrease in S and G2/M fractions. (Fig. 6B)
Figure 6. H-89 shows dose dependent inhibition of proliferation induces cell death and causes G1/S phase arrest in cell cycle.
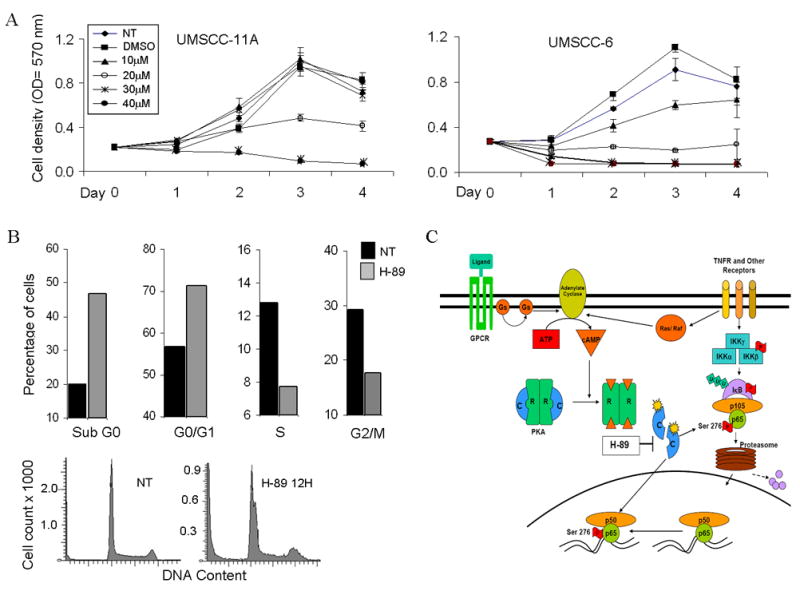
(A) UMSCC-11A and UMSCC-6 cells were treated with different concentrations (10 to 40μm) and proliferation of cells measured using the MTT assay for 4 days after addition of drug. Optical density values were plotted on the X axis for the corresponding days. No treatment and DMSO treated samples were run as controls for comparison. Values shown are an average of three independent observations. (B) DNA cell cycle analysis following treatment of UMSCC-6 cells with 20μM H-89 for 12 hours. Cells were counted and stained with propidium iodide (PI) and cell cycle analysis was done on 20,000 events. Graph shows distribution of cells in various phases of the cell cycle expressed as a percentage, compared with no treatment controls at 12 hours. (C) Model for PKA and phospho-ser276 activation in HNSCC. Ligand binding to TNF receptor (TNFR) or G-Protein Coupled Receptors (GPCR) activating stimulatory G-proteins Ras or Gs activates adenylate cyclase causing accumulation of cAMP. Active PKA catalytic subunits (c) are released following cAMP binding to the regulatory subunits. The active catalytic subunits phosphorylate targets both in the cytoplasm and in the nucleus, including p65 which is phosphorylated at the Ser-276 site. This site specific phosphorylation permits complex formation with transcription activating proteins and modulates NF-κB dependent gene expression.
Discussion
In this study, we present evidence that phosphorylation of p65 at Ser 276 is a common event in head and neck tumors, with ~75% of HNSCC samples showing some degree of activation in the nucleus. It is also appears to be an early event in the evolution of cancer as it is seen to be strongly expressed in peritumoral dysplastic epithelium (Fig 1), either due to genetic alterations already present in the epithelium at risk driving the activation of NF-κB, or as a result of stimulation from cytokines in the tumor microenvironment. The nuclear localization of the phosphorylated protein in the adjacent epithelium with no morphological features of malignancy, as opposed to cytoplasmic localization in normal mucosa, suggests that this site specific phosphorylation may represent either potential of premalignant lesions to progress to overt cancer, or response to inflammatory factors, such as TNF-α, in the tumor microenvironment. In an earlier study, over expression of p65 phosphorylated at Ser 536 by immunohistochemistry has been shown to be associated with high grade dysplasia and carcinoma of the tonsils and increasing levels of immunopositivity were statistically significant predictors of survival (34). Additionally, increasing levels of phospho NF-κB expression in tissue with high-grade dysplasia were also significantly related to progression (34). However, antibodies to p65 phospho-ser276 stain fixed paraffin embedded tissue more reliably than those available for phosphor-ser536. These observations make a strong case for further studies to see if the expression levels of phospho p65 (Ser 276) correlate with statistics of locoregional control and survival.
Phosphorylation of p65 at serine 276 is seen to be rapidly achieved in the presence of TNF-α as observed in our cell lines (Fig 2A). This was consistent with observations on U937 human monocyte-like histiocytic lymphoma cells where phosphorylation was seen as early as 30 minutes (20). Variation in the constitutive and TNF-α inducible levels of phosphorylated p65 (Ser 276) was also noted between the two cell lines by western blot (Fig. 2B), similar to the variable levels of nuclear expression seen in human tissue sections on IHC. TNF-α has been shown to be expressed in HNSCC tumors (28), and the data presented here suggest that TNF-α may not only be a significant inducer of NF-κB nuclear translocation, but important in a PKA-mediated p-p65 (Ser 276) modification and functional regulation of prominent cancer genes, as illustrated in the model in Fig. 6C.
Transactivation of p65 has previously been shown to be induced by the G-protein Ras, and Raf kinase (35, Figure 6C). TNF-α has recently been reported to induce NF-κB p65-ser-276 phosphorylation and promote survival of colon epithelial cells, a mechanism important in development of inflammatory bowel cancer, via Raf dependent signaling in mice (36). G-protein coupled receptors may also activate PKA (Fig. 6C), raising the possibility that other ligands and receptors may contribute to PKA and NF-κB activation in HNSCC. Identification of such ligand/receptors contributing to PKA and NF-κB activation may offer more specific targets than PKA for molecular intervention in HNSCC.
Rapid inhibition of phosphorylation of p65 at serine 276 was noted on treating UMSCC cells with PKA inhibitor H-89, as early as 1 hour after treatment. A decrease in whole cell levels of phospho p65 (Ser 276) in both unstimulated and TNF-α stimulated cell systems is noted. In the cells treated with H-89 for 12 hours and activated with TNF-α, a paradoxical increase is noted, probably due to kinetics of degradation and resynthesis of NF-κB inhibitor, IκBα (37). Similar effects are observed at the same time point in nuclear extracts (Fig 3B, lower panel). The levels of total p65 (whole cell and nuclear) and p65 (Ser 536) do not mirror the changes, suggesting that decrease in phosphorylated p65 (Ser 276) levels is due to inhibition of phosphorylation in the nucleus, and not a manifestation of impaired translocation of p65 to the nucleus in response to drug treatment.
Inhibition of phosphorylation of p65 at Ser 276 by PKA inhibition using drug and siRNA was further shown to inhibit transactivation of NF-κB dependent genes without affecting NF-κB DNA binding. Previous reports suggest the mode of activation of NF-κB by phosphorylation at of p65 at Ser 276 to be due to induction of conformal changes on the p65 molecule by way of weakening of interaction between the N-terminus and C-terminal region induced by phosphorylation, thereby creating and additional site for interaction with co activator CBP/p300 (14). Another mechanism of NF-κB activation has been suggested to be via the association of p-pSer 276 and the P-TEFb transcriptional elongation complex, which is an association of CDK-9 and cyclin T1 (38).
Knockdown of PKA using siRNA directed against PKACα significantly decreased the expression of PKACα protein, along with a decrease in the phosphorylation of p65 at Ser 276. However, the inhibition is not very robust compared to inhibition achieved by H-89 (Fig 5B), raising the possibility of a contribution of cross-reactive inhibition of MSK-1 induced phosphorylation of p65 at Ser 276 (15, 16) by H-89. Though H-89 is a very potent inhibitor of PKA, various studies have questioned the selectivity of H-89 as an inhibitor of PKA (18) and recent studies have shown MSK-1 to contribute to the phosphorylation of p65 at serine 276. Contrastingly however, another study on monocytic cells does not identify MSK-1 as being a major kinase controlling serine 276 phosphorylation on p65 (20). Our experiments also failed to show any significant level of inhibition of NF-κB luciferase activity following siRNA knockdown of MSK-1.
Figure 5. H-89 modulates expression of genes previously implicated in the malignant phenotype of HNSCC.

UMSCC cells were treated with 20 μM H-89 for varying durations as shown and RNA extracted and reverse transcribed. Quantitative RT-PCR expression levels for all genes illustrated here were normalized to no treatment controls and expressed on a log scale. Each value is representative of three independent observations. (p values shown on the graphs represent the significance of highest knockdown achieved during the time points measured compared to no treatment controls.)
Our demonstration of the decrease in expression levels of Cyclin D1 (CCND1) at an early time point and a corresponding increase in CDK inhibitor p21 is also reflected by the decrease in proliferation as seen by decreased expression of Ki-67 at later time points. Our results are consistent with observations that NF-κB mediates cell cycle progression through direct binding of the cyclin D1 promoter at multiple sites and regulates progression through the G1-S cell cycle checkpoint (39). NF-κB has also been shown to inhibit apoptosis by directly binding the promoter and inducing genes encoding BCL-2 homologue BCL-XL (40), and decreased mRNA expression of anti apoptotic genes BCL-2 and BCL-XL highlighted the contribution of downregulation of these genes to the induction of cell death by H-89 as shown by flow cytometry. Potential for inhibitory effects on angiogenesis were demonstrated by inhibition of expression of VEGF. VEGF has been shown to be highly overexpressed in head and neck cancer and increased levels in the serum have been identified as a markers for poor prognosis in head and neck cancer (27, 41). COX-2 and IL-8 are the other NF-κB downstream genes associated with inflammation, angiogenesis and the malignant phenotype in HNSCC that were significantly inhibited by H-89.
Our experiments showed that H-89 induced PKA inhibition had pronounced cytotoxic and growth inhibitory effects on head and neck cell lines as evidenced by MTT assays done of UMSCC-6 and UMSCC-11 A. Perturbations on cell cycle noted on drug treatment included accumulation of cells in the G0/G1 phase with subsequent decrease in the S phase, consistent with the inhibition of cyclin D1 by H-89 (42). This is further reinforced by a significant decrease in cyclin D1 mRNA levels seen in drug treated cells on RT-PCR (Fig 6). The mode of cell cycle arrest induced by H-89 treatment has been reported to be a result of PKA mediated formation of an insoluble complex of cyclin D1 protein in the nuclear matrix structures (42). Cytotoxic effects of the drug was also evident at 12 hours of drug treatment, shown by an increase in the sub-G0 population of treated cells (Fig 4B).
Strategies to inhibit PKA activation in the prevention and development of cancers are under investigation. Antisense oligonucleotides to the regulatory subunit of PKA have shown significant chemopreventive effects on the development of tumors in a DMBA induced mammary carcinogenesis model (43). Antisense oligonucleotides that reduce PKA expression have also been shown to be effective in breast, colorectal and gastric carcinomas in vitro (44) and sensitized colorectal and ovarian cancer cell lines to radiotherapy (45) and chemotherapy in vivo (46). Supporting the potential of PKA or its target phosphorylation as a biomarker, overexpressed PKA was also studied in a subset of patients enrolled for RTOG Trial 86–10 for prostate cancer. Multivariate analysis showed a significant relationship between overexpressed PKA and local failure, biochemical failure and distant metastasis (47).
Inhibition of p65 phosphorylation is also being recognized as an important strategy to inhibit tumorigenesis. Anti tumor effects of the drug P3-25[(5-(4-methoxyarylimino)-2-N-(3, 4-dichlorophenyl)-3-oxo-1, 2, 4-thiadiazolidine] were shown to be due to inhibition of upstream kinases PKA and CK2, resulting in impairment of p65 phosphorylation at Ser 536 and Ser 276 (48). Treatment with this drug induced apoptosis in doxorubicin resistant breast cancer cells and potentiated apoptosis in the presence of other chemotherapeutic agents. Interestingly, there is also evidence that NSAIDS like piroxicam, naproxen, nimesulide and acetylsalicylic acid cause inhibition of c-AMP dependent activation of PKA and in a COX- independent manner (49, 50). These findings raise the potential of combination of NSAIDS alone or with canonical NF-κB p-p65 (Ser 536) inhibitors in future chemoprevention trials.
Here, we have, for the first time, shown that a PKA mediated phosphorylation of p65 at serine 276 an important event in the constitutive and TNF induced activation of NF-κB in HNSCC cell lines and primary tumor. This site specific phosphorylation was found to be important for NF-κB transactivation and has been shown to regulate expression of genes controlling various components of the cancer phenotype including the cell cycle, apoptosis, inflammation, angiogenesis and proliferation. The study highlights the potential and important role of kinases other than IKK to activate the NF-κB signaling cascade and highlights the fact that inhibition of these kinases or their effects on site specific phosphorylations may need to be addressed in future prevention and treatment strategies. The over-expression of p-p65 (Ser 276) at the tumor margins and in dysplastic epithelium warrant investigation as a biomarker to predict the behavior of premalignant lesions of head and neck and to assess efficacy of chemopreventive interventions.
Supplementary Material
Acknowledgments
The authors wish to thank Dr. Alfredo Molinolo (National Institute of Dental and Craniofacial Research) for providing his tissue array and assistance with immunohistochemistry and Dr. Chris Silvin (Genetics and Molecular Biology Branch, Disorders of Immunity Section, NHGRI/NIH) for technical assistance with Flow Cytometry. Drs. James F. Battey and Silvio Gutkind served as readers and provided helpful comments. Supported by NIDCD intramural project Z01-DC-000016.
Footnotes
Translational Relevance
We discovered previously that aberrant activation of the NF-κB pathway through IKK mediated phosphorylation of REL-A/p65 (Ser 536) is prevalent and important in the malignant phenotype of HNSCC. Here, we provide evidence for a key role of p65 (Ser 276) phosphorylation by Protein Kinase A (PKA) in the constitutive and TNF-α-induced transactivating function of NF-κB, modulation of key genes implicated in the malignant phenotype, and promotion of cell cycle progression and growth in human HNSCC lines. Thus, inhibitors of PKA, TNF-α or other components upstream of this pathway may be targets for chemo-intervention. Furthermore, nuclear p65 (Ser276) phosphorylation is prevalent in HNSCC and dysplastic tissues, in contrast to the cytoplasmic localization in normal mucosa, meriting investigation as an early biomarker for malignant transformation.
Supported by NIDCD Intramural project Z01-DC-000016 and Z01-DC-000073
References
- 1.Chang AA, Van Waes C. Nuclear factor-KappaB as a common target and activator of oncogenes in head and neck squamous cell carcinoma. Adv Otorhinolaryngol. 2005;62:92–102. doi: 10.1159/000082476. [DOI] [PubMed] [Google Scholar]
- 2.Loercher A, Lee TL, Ricker JL, et al. Nuclear factor-kappaB is an important modulator of the altered gene expression profile and malignant phenotype in squamous cell carcinoma. Cancer Res. 2004;64:6511–23. doi: 10.1158/0008-5472.CAN-04-0852. [DOI] [PubMed] [Google Scholar]
- 3.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–83. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 4.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–62. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- 5.Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. 2006;210:171–86. doi: 10.1111/j.0105-2896.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 6.Buss H, Dorrie A, Schmitz ML, et al. Phosphorylation of serine 468 by GSK-3beta negatively regulates basal p65 NF-kappaB activity. J Biol Chem. 2004;279:49571–4. doi: 10.1074/jbc.C400442200. [DOI] [PubMed] [Google Scholar]
- 7.Haller D, Russo MP, Sartor RB, Jobin C. IKK beta and phosphatidylinositol 3-kinase/Akt participate in non-pathogenic Gram-negative enteric bacteria-induced RelA phosphorylation and NF-kappa B activation in both primary and intestinal epithelial cell lines. J Biol Chem. 2002;277:38168–78. doi: 10.1074/jbc.M205737200. [DOI] [PubMed] [Google Scholar]
- 8.Mattioli I, Sebald A, Bucher C, et al. Transient and selective NF-kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J Immunol. 2004;172:6336–44. doi: 10.4049/jimmunol.172.10.6336. [DOI] [PubMed] [Google Scholar]
- 9.Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem. 1999;274:30353–6. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- 10.Sakurai H, Suzuki S, Kawasaki N, et al. Tumor necrosis factor-alpha-induced IKK phosphorylation of NF-kappaB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J Biol Chem. 2003;278:36916–23. doi: 10.1074/jbc.M301598200. [DOI] [PubMed] [Google Scholar]
- 11.Schwabe RF, Sakurai H. IKKbeta phosphorylates p65 at S468 in transactivaton domain 2. FASEB J. 2005;19:1758–60. doi: 10.1096/fj.05-3736fje. [DOI] [PubMed] [Google Scholar]
- 12.Yang F, Tang E, Guan K, Wang CY. IKK beta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J Immunol. 2003;170:5630–5. doi: 10.4049/jimmunol.170.11.5630. [DOI] [PubMed] [Google Scholar]
- 13.Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–71. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]
- 14.Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S. The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell. 1997;89:413–24. doi: 10.1016/s0092-8674(00)80222-6. [DOI] [PubMed] [Google Scholar]
- 15.Vermeulen L, De Wilde G, Van Damme P, Vanden Berghe W, Haegeman G. Transcriptional activation of the NF-kappaB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1) EMBO J. 2003;22:1313–24. doi: 10.1093/emboj/cdg139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joo JH, Jetten AM. NF-kappaB-dependent transcriptional activation in lung carcinoma cells by farnesol involves p65/RelA(Ser276) phosphorylation via the MEK-MSK1 signaling pathway. J Biol Chem. 2008;283:16391–9. doi: 10.1074/jbc.M800945200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson-White AJ, Hsiao HP, Leitner WW, et al. Protein kinase A-independent inhibition of proliferation and induction of apoptosis in human thyroid cancer cells by 8-Cl-adenosine. J Clin Endocrinol Metab. 2008;93:1020–9. doi: 10.1210/jc.2007-2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lochner A, Moolman JA. The many faces of H89: a review. Cardiovasc Drug Rev. 2006;24:261–74. doi: 10.1111/j.1527-3466.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- 19.Cho-Chung YS. Role of cyclic AMP receptor proteins in growth, differentiation, and suppression of malignancy: new approaches to therapy. Cancer Res. 1990;50:7093–100. [PubMed] [Google Scholar]
- 20.Jamaluddin M, Wang S, Boldogh I, Tian B, Brasier AR. TNF-alpha-induced NF-kappaB/RelA Ser(276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell Signal. 2007;19:1419–33. doi: 10.1016/j.cellsig.2007.01.020. [DOI] [PubMed] [Google Scholar]
- 21.Lu SL, Herrington H, Reh D, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes Dev. 2006;20:1331–42. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel V, Rosenfeldt HM, Lyons R, et al. Persistent activation of Rac1 in squamous carcinomas of the head and neck: evidence for an EGFR/Vav2 signaling axis involved in cell invasion. Carcinogenesis. 2007;28:1145–52. doi: 10.1093/carcin/bgm008. [DOI] [PubMed] [Google Scholar]
- 23.Vitale-Cross L, Amornphimoltham P, Fisher G, Molinolo AA, Gutkind JS. Conditional expression of K-ras in an epithelial compartment that includes the stem cells is sufficient to promote squamous cell carcinogenesis. Cancer Res. 2004;64:8804–7. doi: 10.1158/0008-5472.CAN-04-2623. [DOI] [PubMed] [Google Scholar]
- 24.Thomas SM, Bhola NE, Zhang Q, et al. Cross-talk between G protein-coupled receptor and epidermal growth factor receptor signaling pathways contributes to growth and invasion of head and neck squamous cell carcinoma. Cancer Res. 2006;66:11831–9. doi: 10.1158/0008-5472.CAN-06-2876. [DOI] [PubMed] [Google Scholar]
- 25.Ondrey FG, Dong G, Sunwoo J, et al. Constitutive activation of transcription factors NF-(kappa)B, AP-1, and NF-IL6 in human head and neck squamous cell carcinoma cell lines that express pro-inflammatory and pro-angiogenic cytokines. Mol Carcinog. 1999;26:119–29. doi: 10.1002/(sici)1098-2744(199910)26:2<119::aid-mc6>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 26.Yu M, Yeh J, Van Waes C. Protein kinase casein kinase 2 mediates inhibitor-kappaB and aberreant NF-kappaB activation by serum factor(s) in head and neck squamous cell carcinoma cells. Cancer Res. 2006;66:6722–31. doi: 10.1158/0008-5472.CAN-05-3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Z, Malhotra PS, Thomas GR, et al. Expression of proinflammatory and proangiogenic cytokines in patients with head and neck cancer. Clin Cancer Res. 1999;5:1369–79. [PubMed] [Google Scholar]
- 28.Younes F, Quartey EL, Kiguwa S, Partridge M. Expression of TNF and the 55-kDa TNF receptor in epidermis, oral mucosa, lichen planus and squamous cell carcinoma. Oral Dis. 1996;2:25–31. doi: 10.1111/j.1601-0825.1996.tb00199.x. [DOI] [PubMed] [Google Scholar]
- 29.Sung MH, Bagain L, Chen Z, et al. Dynamic effect of bortezomib on nuclear factor-kappaB activity and gene expression in tumor cells. Mol Pharmacol. 2008;74:1215–22. doi: 10.1124/mol.108.049114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duan J, Friedman J, Nottingham L, Chen Z, Ara G, Van Waes C. Nuclear factor-kappaB p65 small interfering RNA or proteasome inhibitor Bortezomib sensitizes head and neck squamous cell carcinomas to classic histone deacetylase inhibitors and novel histone deacetylase inhibitor PXD101. Mol Cancer Ther. 2007;6:37–50. doi: 10.1158/1535-7163.MCT-05-0285. [DOI] [PubMed] [Google Scholar]
- 31.Chan G, Boyle JO, Yang EK, et al. Cyclooxygenase-2 expression is up-regulated in squamous cell carcinoma of the head and neck. Cancer Res. 1999;59:991–4. [PubMed] [Google Scholar]
- 32.Mestre JR, Chan G, Zhang F, et al. Inhibition of cyclooxygenase-2 expression. An approach to preventing head and neck cancer. Ann N Y Acad Sci. 1999;889:62–71. doi: 10.1111/j.1749-6632.1999.tb08724.x. [DOI] [PubMed] [Google Scholar]
- 33.Lin DT, Subbaramaiah K, Shah JP, Dannenberg AJ, Boyle JO. Cyclooxygenase-2: a novel molecular target for the prevention and treatment of head and neck cancer. Head Neck. 2002;24:792–9. doi: 10.1002/hed.10108. [DOI] [PubMed] [Google Scholar]
- 34.Zhang PL, Pellitteri PK, Law A, et al. Overexpression of phosphorylated nuclear factor-kappa B in tonsillar squamous cell carcinoma and high-grade dysplasia is associated with poor prognosis. Mod Pathol. 2005;18:924–32. doi: 10.1038/modpathol.3800372. [DOI] [PubMed] [Google Scholar]
- 35.Finco TS, Westwick JK, Norris JL, Beg AA, Der CJ, Baldwin AS., Jr Oncogenic Ha-Ras-induced signaling activates NF-kappaB transcriptional activity, which is required for cellular transformation. J Biol Chem. 1997;272:24113–6. doi: 10.1074/jbc.272.39.24113. [DOI] [PubMed] [Google Scholar]
- 36.Edelblum KL, Goettel JA, Koyama T, McElroy SJ, Yan F, Polk DB. TNFR1 promotes tumor necrosis factor-mediated mouse colon epithelial cell survival through RAF activation of NF-kappaB. J Biol Chem. 2008;283:29485–94. doi: 10.1074/jbc.M801269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nelson DE, Ihekwaba AE, Elliott M, et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. 2004;306:704–8. doi: 10.1126/science.1099962. [DOI] [PubMed] [Google Scholar]
- 38.Nowak DE, Tian B, Jamaluddin M, et al. RelA Ser276 phosphorylation is required for activation of a subset of NF-kappaB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol Cell Biol. 2008;28:3623–38. doi: 10.1128/MCB.01152-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS., Jr NF-kappaB controls growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–99. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zong WX, Edelstein LC, Chen C, Bash J, Gelinas C. The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kappaB that blocks TNFalpha-induced apoptosis. Genes Dev. 1999;13:382–7. doi: 10.1101/gad.13.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allen C, Duffy S, Teknos T, et al. Nuclear factor-kappaB-related serum factors as longitudinal biomarkers of response and survival in advanced oropharyngeal cancer. Clin Cancer Res. 2007;13:3182–90. doi: 10.1158/1078-0432.CCR-06-3047. [DOI] [PubMed] [Google Scholar]
- 42.Scovassi AI, Stivala LA, Rossi L, Bianchi L, Prosperi E. Nuclear association of cyclin D1 in human fibroblasts: tight binding to nuclear structures and modulation by protein kinase inhibitors. Exp Cell Res. 1997;237:127–34. doi: 10.1006/excr.1997.3770. [DOI] [PubMed] [Google Scholar]
- 43.Nesterova MV, Cho-Chung YS. Chemoprevention with protein kinase A RIalpha antisense in DMBA-mammary carcinogenesis. Ann N Y Acad Sci. 2005;1058:255–64. doi: 10.1196/annals.1359.038. [DOI] [PubMed] [Google Scholar]
- 44.Yokozaki H, Budillon A, Tortora G, et al. An antisense oligodeoxynucleotide that depletes RI alpha subunit of cyclic AMP-dependent protein kinase induces growth inhibition in human cancer cells. Cancer Res. 1993;53:868–72. [PubMed] [Google Scholar]
- 45.Bianco C, Bianco R, Tortora G, et al. Antitumor activity of combined treatment of human cancer cells with ionizing radiation and anti-epidermal growth factor receptor monoclonal antibody C225 plus type I protein kinase A antisense oligonucleotide. Clin Cancer Res. 2000;6:4343–50. [PubMed] [Google Scholar]
- 46.Agrawal S, Kandimalla ER, Yu D, et al. GEM 231, a second-generation antisense agent complementary to protein kinase A RIalpha subunit, potentiates antitumor activity of irinotecan in human colon, pancreas, prostate and lung cancer xenografts. Int J Oncol. 2002;21:65–72. [PubMed] [Google Scholar]
- 47.Khor LY, Bae K, Al-Saleem T, et al. Protein kinase A RI-alpha predicts for prostate cancer outcome: analysis of radiation therapy oncology group trial 86–10. Int J Radiat Oncol Biol Phys. 2008;71:1309–15. doi: 10.1016/j.ijrobp.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manna SK, Manna P, Sarkar A. Inhibition of RelA phosphorylation sensitizes apoptosis in constitutive NF-kappaB-expressing and chemoresistant cells. Cell Death Differ. 2007;14:158–70. doi: 10.1038/sj.cdd.4401929. [DOI] [PubMed] [Google Scholar]
- 49.Zentella de Pina M, Vazquez-Meza H, Agundis C, et al. Inhibition of cAMP-dependent protein kinase A: a novel cyclo-oxygenase-independent effect of non-steroidal anti-inflammatory drugs in adipocytes. Auton Autacoid Pharmacol. 2007;27:85–92. doi: 10.1111/j.1474-8673.2007.00392.x. [DOI] [PubMed] [Google Scholar]
- 50.Bock JM, Menon SG, Goswami PC, et al. Relative non-steroidal anti-inflammatory drug (NSAID) antiproliferative activity is mediated through p21-induced G1 arrest and E2F inhibition. Mol Carcinog. 2007;46:857–64. doi: 10.1002/mc.20318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.