
Abstract
Three human coronaviruses are known to exist: human coronavirus 229E (HCoV-229E), HCoV-OC43 and severe acute respiratory syndrome (SARS)-associated coronavirus (SARS-CoV). Here we report the identification of a fourth human coronavirus, HCoV-NL63, using a new method of virus discovery. The virus was isolated from a 7-month-old child suffering from bronchiolitis and conjunctivitis. The complete genome sequence indicates that this virus is not a recombinant, but rather a new group 1 coronavirus. The in vitro host cell range of HCoV-NL63 is notable because it replicates on tertiary monkey kidney cells and the monkey kidney LLC-MK2 cell line. The viral genome contains distinctive features, including a unique N-terminal fragment within the spike protein. Screening of clinical specimens from individuals suffering from respiratory illness identified seven additional HCoV-NL63-infected individuals, indicating that the virus was widely spread within the human population.
Similar content being viewed by others
Main
To date, there is still a variety of human diseases with unknown etiology. A viral origin has been suggested for many of these diseases, emphasizing the importance of a continuous search for new viruses1,2,3. Major difficulties are encountered, however, when searching for new viruses. First, some viruses do not replicate in vitro, at least not in the cells that are commonly used in viral diagnostics. Second, for those viruses that do replicate in vitro and cause a cytopathic effect (CPE), the subsequent virus identification methods may fail. Antibodies raised against known viruses may not recognize the cultured virus, and virus-specific PCR methods may not amplify the new viral genome. To solve both problems, we developed a new method for virus discovery based on the cDNA-amplified restriction fragment–length polymorphism technique (cDNA-AFLP4). Here we report the identification of a new coronavirus using this method of Virus-Discovery-cDNA-AFLP (VIDISCA).
Coronaviruses, a genus of the Coronaviridae family, are enveloped viruses with a large plus-strand RNA genome. The genomic RNA is 27–32 kb in size, capped and polyadenylated. Three serologically distinct groups of coronaviruses have been described. Within each group, viruses are characterized by their host range and genome sequence. Coronaviruses have been identified in mice, rats, chickens, turkeys, swine, dogs, cats, rabbits, horses, cattle and humans, and can cause a variety of severe diseases including gastroenteritis and respiratory tract diseases5,6. Three human coronaviruses have been studied in detail. HCoV-229E and HCoV-OC43 were identified in the mid-1960s, and are known to cause the common cold7,8,9,10,11,12,13,14,15. The recently identified SARS-CoV causes a life-threatening pneumonia, and is the most pathogenic human coronavirus identified thus far16,17,18. SARS-CoV is likely to reside in an animal reservoir, and has recently initiated the epidemic in humans through zoonotic transmission19,20. It has been suggested that SARS-CoV is the first member of a fourth group of coronaviruses, or that it is an outlier of group 2 (refs. 21, 22).
The new coronavirus that we present here was isolated from a child suffering from bronchiolitis and conjunctivitis. This was not an isolated case, as we identified the virus in clinical specimens from seven additional individuals, both infants and adults, during the last winter season. We also resolved the complete sequence of the viral genome, which revealed several unique features.
Results
Virus isolation from a child with acute respiratory disease
In January 2003, a 7-month-old child was admitted to the hospital with coryza, conjunctivitis and fever. Chest radiography revealed typical features of bronchiolitis. A nasopharyngeal aspirate specimen was collected 5 d after the onset of disease (sample NL63). Diagnostic tests for respiratory syncytial virus, adenovirus, influenza viruses A and B, parainfluenza virus types 1, 2 and 3, rhinovirus, enterovirus, HCoV-229E and HCoV-OC43 yielded negative results. The clinical sample was subsequently inoculated onto human fetal lung fibroblasts, tertiary monkey kidney cells (Cynomolgus monkey) and HeLa cells. CPE was detected exclusively on tertiary monkey kidney cells, and was first noted 8 d after inoculation. The CPE was diffuse, with a refractive appearance in the affected cells followed by cell detachment. More pronounced CPE was observed upon passage onto the monkey kidney cell line LLC-MK2, with overall cell rounding and moderate cell enlargement (Supplementary Fig. 1 online). Additional subcultures on human fetal lung fibroblasts, rhabdomyosarcoma cells and Vero cells remained negative for CPE. Immunofluorescence assays to detect respiratory syncytial virus, adenovirus, influenza viruses A and B, and parainfluenza virus types 1, 2 and 3 remained negative. Acid lability and chloroform sensitivity tests indicated that the virus was most likely enveloped, and did not belong to the picornavirus group23.
Virus discovery by the VIDISCA method
Identification of unknown pathogens using molecular biology tools is difficult because the target sequence is not known, so genome-specific PCR primers cannot be designed. To overcome this problem, we developed the VIDISCA method based on the cDNA-AFLP technique4. The advantage of VIDISCA is that prior knowledge of the sequence is not required, as the presence of restriction enzyme sites is sufficient to guarantee PCR amplification. The input sample can be either blood plasma or serum, or culture supernatant. Whereas cDNA-AFLP starts with isolated mRNA, VIDISCA begins with a treatment to selectively enrich for viral nucleic acid, including a centrifugation step to remove residual cells and mitochondria (Fig. 1a). A DNase treatment is also used to remove interfering chromosomal and mitochondrial DNA from degraded cells (viral nucleic acid is protected within the viral particle). Finally, by choosing frequently cutting restriction enzymes, the method can be fine-tuned such that most viruses will be amplified. We were able to amplify viral nucleic acids in EDTA-treated plasma from a person with hepatitis B viral infection, and from a person with an acute parvovirus B19 infection (Fig. 1b). The technique can also detect HIV-1 in cell culture, demonstrating its capacity to identify both RNA and DNA viruses (Fig. 1b).

(a) Schematic overview of steps in VIDISCA method. (b) Examples of VIDISCA-mediated virus identification. Specimens were analyzed using ethidium bromide–stained agarose (parvovirus B19) or Metaphor agarose (HBV and HIV-1) gel electrophoresis. Lane M, DNA molecular weight markers; −, negative controls; +, VIDISCA PCR products for HBV (amplified with primers HinP1I-T/MseI-T), parvovirus B19 (HinP1I standard primer only) or HIV-1 (EcoRI-A/MseI-C primers). (c) VIDISCA PCR products for HCoV-NL63. HinP1I-G and MseI-A primers were used for selective amplification; products were visualized by Metaphor agarose gel electrophoresis. Lanes 1 and 2, duplicate PCR product of cultured HCoV-NL63 harvested from LLC-MK2 cells; 3 and 4, duplicate control supernatant from uninfected LLC-MK2 cells; 5 and 6, duplicate negative controls containing water; M, 25-bp molecular weight marker. Arrow indicates HCoV-NL63 fragment that was excised from gel and sequenced.
The supernatant of the CPE-positive LLC-MK2 culture NL63 was analyzed by VIDISCA. The supernatant of uninfected cells was used as a negative control. After the second PCR amplification step, unique and prominent DNA fragments were present in the test sample but not in the control (1 of 16 selective PCR reactions is shown in Fig. 1c). These fragments were cloned and sequenced. Thirteen of 16 fragments showed sequence similarity to members of the coronavirus family, but significant sequence divergence with known coronaviruses was apparent in all fragments, indicating that we had identified a new coronavirus. The sequences of the 13 VIDISCA fragments are provided in Supplementary Figure 2 online.
Detection of HCoV-NL63 in patient specimens
To show that HCoV-NL63 originated from the nasopharyngeal aspirate of the child, we designed a diagnostic RT-PCR that specifically detects HCoV-NL63. This test confirmed the presence of HCoV-NL63 in the clinical sample. The sequence of the RT-PCR product of the 1b gene was identical to that of the virus identified upon in vitro passage in LLC-MK2 cells (data not shown).
Having confirmed that the cultured coronavirus originated from the child, the question remained as to whether this was an isolated clinical case, or whether HCoV-NL63 is circulating in humans. To address this question, we used two diagnostic RT-PCR assays to examine respiratory specimens of hospitalized individuals and those visiting the outpatient clinic between December 2002 and August 2003 (Fig. 2). We identified seven additional individuals carrying HCoV-NL63 (Table 1). Sequence analysis of the PCR products indicated the presence of a few characteristic point mutations in several samples, suggesting that several viruses with different molecular markers may be cocirculating (Fig. 3 and Supplementary Fig. 3 online). At least five of the HCoV-NL63-positive individuals suffered from respiratory tract illness; the clinical data of two individuals was not available. Including the index case, five of the patients were children less than 1 year old, and three patients were adults. Two adults were likely to be immunosuppressed, as one of them was a bone marrow transplant recipient and the other an HIV-positive patient suffering from AIDS, with very low CD4+ cell counts (Table 1). No clinical data was available for the third adult. One patient was coinfected with respiratory syncytial virus (no. 72), and the HIV-infected patient (no. 466) carried Pneumocystis carinii. No other respiratory agent was found in the other patients, suggesting that the respiratory symptoms were caused by HCoV-NL63. All positive samples were collected during the last winter season, with a detection frequency of 7% in January 2003. None of the 306 samples collected in the spring and summer of 2003 contained HCoV-NL63 (P < 0.01 by two-tailed t test).

(a) Number of patients tested per month. (b) Percentage of patients positive for HCoV-NL63.

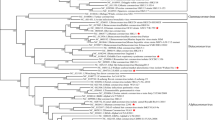
HCoV-229E was used to root the tree.
Complete genome analysis of HCoV-NL63
The genomes of coronaviruses have a characteristic organization. The 5′ two-thirds contain the 1a and 1b genes that encode the nonstructural polyproteins, followed by the genes encoding four structural proteins: spike (S), envelope (E), membrane (M) and nucleocapsid (N). The genomes of known coronaviruses contain a variable number of unique characteristic open reading frames (ORFs) encoding nonstructural proteins either between the 1b and S genes, between the S and E genes, between the M and N genes, or downstream of the N gene.
To determine whether the HCoV-NL63 genome organization shares these characteristics, we constructed a cDNA library with purified virus stock as input material. A total of 475 genome fragments were analyzed, with an average coverage of seven sequences per nucleotide. Specific PCR reactions were designed to fill in gaps and to sequence regions with low-quality sequence data. We combined this with 5′ and 3′ rapid amplification of cDNA ends to resolve the complete HCoV-NL63 genome sequence.
The RNA genome of HCoV-NL63 consists of 27,553 nucleotides and a poly-A tail. With a GC content of 34%, HCoV-NL63 has the lowest GC content among the Coronaviridae, which range from 37–42% (ref. 24). ZCurve software was used to identify the ORFs25, and the genome configuration was portrayed using the similarity with known coronaviruses as a guide (Fig. 4a and Supplementary Table 1 online). Short untranslated regions (UTRs) of 286 and 287 nucleotides are present at the 5′ and 3′ termini, respectively. The 1a and 1b genes encode the RNA polymerase and proteases that are essential for virus replication. A potential pseudoknot structure is present at position 12,439 (data not shown), which may provide the −1 frameshift signal to translate the 1b polyprotein. Genes predicted to encode the S, E, M and N proteins are found in the 3′ part of the genome. The hemagglutinin-esterase gene, which is present in some group 2 coronaviruses, is not present in HCoV-NL63. ORF3, located between the S and E genes, probably encodes a single accessory nonstructural protein; this gene showed only limited similarity to ORF4A and ORF4B of HCoV-229E and ORF3 of porcine epidemic diarrhea virus (PEDV).

(a) ORFs encoding 1a, 1b, S, ORF3, E, M and N proteins are flanked by 286-nucleotide 5′ UTR and 287-nucleotide 3′ UTR. Coordinates of each ORF are provided in Supplementary Table 1 online. (b) Phylogenetic analysis of HCoV-NL63, using nucleotide sequences predicted to encode 1a, 1b, S, M and N proteins (see Supplementary Methods online for GenBank accession numbers). Red, group 1 viruses; blue, group 2; green, group 3; purple, SARS-CoV. MHV, mouse hepatitis virus; IBV, avian infectious bronchitis virus; BCoV, bovine coronavirus; FCoV, feline enteric coronavirus; CCoV, canine coronavirus; FIPV, feline infectious peritonitis virus; EqCoV, equine coronavirus; TCoV, turkey coronavirus.
The 1a and 1ab polyproteins are translated from the genomic RNA, but the remaining viral proteins are translated from subgenomic mRNAs made by discontinuous transcription during negative strand synthesis26. Each subgenomic mRNA has a common 5′ end, derived from the 5′ portion of the genome (the 5′ leader sequence), and common 3′ coterminal parts. Discontinuous transcription requires base-pairing between cis-acting transcription regulatory sequences (TRSs), one located near the 5′ part of the viral genome (the leader TRS) and others located upstream of each of the respective ORFs (the body TRSs)27. The cDNA bank that we sequenced contained copies of the subgenomic mRNA for the N protein, thus providing the opportunity to exactly map the leader sequence that is fused to all subgenomic mRNAs. A leader of 72 nucleotides was identified at the 5′ UTR. Eleven of twelve nucleotides of the leader TRS (5′-UCUCAACUAAAC-3′) showed similarity with the body TRS upstream of the N gene. Putative TRSs were also identified upstream of the S, ORF3, E and M genes (Supplementary Table 2 online).
We next aligned the sequence of HCoV-NL63 with the complete genomes of other coronaviruses. The percentage nucleotide identity was determined for each gene and is listed in Table 2. All genes except the M gene shared the highest identity with HCoV-229E. To confirm that HCoV-NL63 is a new member of the group 1 coronaviruses, we conducted phylogenetic analysis using the nucleotide sequence of the 1a, 1b, S, M and N genes (Fig. 4b). For each gene analyzed, HCoV-NL63 clustered with the group 1 coronaviruses. The 1a, 1b and S genes of HCoV-NL63 are most closely related to those of HCoV-229E. However, further inspection revealed a subcluster of HCoV-NL63, HCoV-229E and PEDV. Phylogenetic analysis could not be performed for the ORF3 and E genes because the regions were too variable or too small for analysis, respectively. Bootscan analysis by the Simplot software version 2.5 (ref. 28) found no signs of recombination (data not shown).
The presence of a single nonstructural gene between the S and E genes is noteworthy because almost all coronaviruses have two or more ORFs in this region, with the exception of PEDV and HCoV-OC43 (ref. 29,30). Perhaps most notable is a large insert of 537 nucleotides in the 5′ portion of the S gene of HcoV-NL63, as compared with that of HCoV-229E. A BLAST search found no similarity between this additional 179–amino acid domain of the S protein and any coronavirus or other sequence deposited in GenBank. An alignment of the HCoV-NL63 S protein sequence with those of other group 1 coronaviruses is shown in Supplementary Figure 4 online.
Discussion
In this study we present a detailed description of a new human coronavirus. Thus far, only three human coronaviruses have been characterized if we include SARS-CoV; further characterization of HCoV-NL63 as the fourth member will provide important insight into the variation among human coronaviruses. HCoV-NL63 is a member of the group 1 coronaviruses and is most closely related to HCoV-229E, but the differences between them are prominent. First, they share on average only 65% sequence identity. Second, a single gene, ORF3, in HCoV-NL63 takes the place of the 4A and 4B genes of HCoV-229E. Third, the 5′ region of the S gene of HCoV-NL63 contains a large in-frame insertion of 537 nucleotides. The N-terminal region of the S protein has been implicated in binding to aminopeptidase N (group I coronaviruses) and sialic acid31,32,33, so the 179–amino acid insert in HCoV-NL63 might be involved in receptor binding and may explain the tropism of this virus in cell culture. However, the aminopeptidase N receptor-binding domain of the HCoV-229E S protein has been mapped to amino acids (407–547 ref. 33), so it seems unlikely that the insertion will be directly involved in binding to aminopeptidase N. Fourth, whereas HCoV-229E is fastidious in cell culture with a narrow host range, HCoV-NL63 replicates efficiently in monkey kidney cells. SARS-CoV is also able to replicate in monkey kidney cells (Vero-E6 cells34), yet the predicted S proteins of SARS-CoV and HCoV-NL63 do not share a domain that could explain the in vitro host cell range of these viruses. Other viral proteins may influence the cell tropism of a virus, but none of the HCoV-NL63 proteins were more closely related to SARS-CoV than to HCoV-229E.
Variability at the 5′ end of the S gene, correlating with alterations in tropism, has also been described for the group 1 coronaviruses porcine respiratory coronavirus (PRCoV) and transmissible gastroenteritis virus (TGEV). These porcine viruses are antigenically and genetically related, but their pathogenicity is markedly different. TGEV replicates in and destroys the enterocytes of the small intestine, causing severe diarrhea with high mortality in neonatal swine. In contrast, PRCoV (which emerged more recently than TGEV) has a selective tropism for respiratory tissue, and very little capacity to replicate in intestinal tissue. The difference between the TGEV and PRCoV S gene sequences is comparable to the difference between those of HCoV-NL63 and HCoV-229E35. Compared with TGEV, PRCoV contains a deletion in the 5′ hypervariable region of the S gene. The extra region that is present at the 5′ end of the TGEV S gene is responsible for the hemagglutination activity of TGEV, and its capacity to bind to sialic acid32. However, this region shows no similarity to the HCoV-NL63 insert.
The common cold–causing virus HCoV-229E can cause more serious respiratory disease in infants and immunocompromised patients36,37. Our data indicate that HCoV-NL63 causes acute respiratory disease in children below the age of 1 year, and in immunocompromised adults. To date, no known viral pathogen can be identified in a substantial portion of respiratory disease cases in humans (20–30%; ref. 38). Several assays have been used to diagnose coronavirus infections. Traditionally, an antibody test is implemented to measure a rise in titers of antibodies to the human coronaviruses HCoV-229E or HCoV-OC43 (ref. 12). Antibodies to HCoV-NL63 might cross-react with HCoV-229E, given that these viruses are members of the same serotype. If this were the case, HCoV-NL63 infections might have been misdiagnosed as HCoV-229E. Molecular biology tools such as RT-PCR assays39,40 were designed to selectively detect the human coronaviruses HCoV-229E and HCoV-OC43, but these assays will not detect HCoV-NL63. Even the RT-PCR assay that was designed to amplify all known coronaviruses40 is not able to amplify HCoV-NL63 because of several mismatches with the primer sequences. The availability of the complete HCoV-NL63 genome sequence means that these diagnostic assays can be substantially improved.
Our results indicate that HCoV-NL63 is present in a significant number of respiratory tract illnesses of unknown etiology. HCoV-NL63 was detected in patients suffering from respiratory disease, with a frequency of up to 7% in January 2003. The virus was not detected in more recent samples collected in the spring and summer of 2003, which correlates with the fact that human coronaviruses tend to be transmitted predominantly in the winter season12. Future experiments with more sensitive diagnostic tools should yield a more accurate picture of the prevalence of this virus and its association with respiratory disease.
Methods
VIDISCA method.
The virus was cultured on LLC-MK2 cells. Details of virus culture and patient descriptions are available in Supplementary Methods online. To remove residual cells and mitochondria, 110 μl of virus culture supernatant was spun for 10 min at maximum speed (13,500 r.p.m.) in an Eppendorf microcentrifuge. To remove chromosomal DNA and mitochondrial DNA from the lysed cells, 100 μl of supernatant was transferred to a fresh tube and treated with DNase I for 45 min at 37 °C (Ambion). Nucleic acids were extracted as described41. A reverse transcription reaction was performed with random hexamer primers (Amersham Bioscience) and Moloney murine leukemia virus reverse transcriptase (MMLV-RT; Invitrogen). Second-strand DNA synthesis was carried out with Sequenase II (Amersham Bioscience), without further addition of a primer. A phenol-chloroform extraction was followed by ethanol precipitation.
cDNA-AFLP was performed essentially as described4, with some modifications. The double-stranded DNA was digested with the HinP1I and MseI restriction enzymes (New England Biolabs). MseI and HinP1I anchors (see below) were subsequently added, along with 5U ligase enzyme (Invitrogen) in the supplied ligase buffer, for 2 h at 37 °C. The MseI and HinP1I anchors were prepared by mixing a top-strand oligonucleotide (5′-CTCGTAGACTGCGTACC-3′ for the MseI anchor and 5′-GACGATGAGTCCTGAC-3′ for the HinP1I anchor) with a bottom-strand oligonucleotide (5′-TAGGTACGCAGTC-3′ for the MseI anchor and 5′-CGGTCAGGACTCAT-3′ for the HinP1I anchor) in a 1:40 dilution of ligase buffer. Twenty cycles of PCR were carried out with 10 μl of the ligation mixture, 100 ng of HinP1I standard primer (5′-GACGATGAGTCCTGACCGC-3′) and 100 ng of MseI standard primer (5′-CTCGTAGACTGCGTACCTAA-3′). Five microliters of this PCR product was used as input in the second 'selective' amplification step, along with 100 ng HinP1I N-primer and 100 ng MseI N-primer (the 'N' indicates that the standard primers were extended with one nucleotide; G, A, T or C). The selective rounds of amplification were done using 'touchdown PCR': 10 cycles of 94 °C for 60 s, 65 °C for 30 s, and 72 °C for 1 min (annealing temperature reduced by 1 °C per cycle); 23 cycles of 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 1 min; and finally 1 cycle of 72 °C for 10 min. Sixteen PCR reactions, each with 1 of the 16 primer combinations, were conducted for each sample in this selective PCR. The PCR products were analyzed on 4% Metaphor agarose gels (Cambrex), and the fragments of interest were cloned and sequenced using BigDye terminator reagents. Electrophoresis and data collection were performed using an ABI 377 instrument. DNA molecular weight markers were from Invitrogen and Eurogentec.
To detect HIV-1, we used VIDISCA with EcoRI digestion instead of HinP1I digestion. VIDISCA was modified for parvovirus B19 detection as follows: the reverse transcription step was excluded; only HinP1I digestion and adaptor ligation were performed; the first PCR reaction was performed with 35 cycles instead of 20; and the first PCR fragments were visualized by agarose gel electrophoresis. Details of cDNA library construction and full genome sequencing are available in Supplementary Methods online.
Diagnostic RT-PCR.
A total of 614 respiratory samples were collected from 493 individuals between December 2002 and August 2003 at the Academic Medical Center in Amsterdam. The specimens included oral and nasopharyngeal aspirates, throat swabs, bronchoalveolar lavage and sputum. The samples had been collected for routine viral diagnostic screening of people suffering from upper and/or lower respiratory tract diseases, and the patients consented that their samples be used for testing of respiratory viruses that included coronaviruses. We used 100 μl of each sample in a Boom extraction41. The diagnostic assay was designed based on the sequence of the 1b gene. The reverse transcription was performed with MMLV-RT (Invitrogen), using 10 ng of reverse transcription primer (repSZ-RT, 5′-CCACTATAAC-3′; coordinate 16232 in HCoV-NL63). The entire reverse transcription mixture was added to the first PCR mixture containing 100 ng of primer repSZ-1 (5′-GTGATGCATATGCTAATTTG-3′; coordinate 15973) and 100 ng of primer repSZ-3 (5′-CTCTTGCAGGTATAATCCTA-3′; coordinate 16210). The PCR reaction consisted of the following steps: 95 °C for 5 min; then 35 cycles of 95 °C for 1 min, 55 °C for 1 min, and 72 °C for 2 min; then 72 °C for 10 min.
A nested PCR was started using 5 μl of the first PCR product with 100 ng of primer repSZ-2 (5′-TTGGTAAACAAAAGATAACT-3′; coordinate 16012) and 100 ng of primer repSZ-4 (5′-TCAATGCTATAAACAGTCAT-3′; coordinate 16181). Twenty-five PCR cycles were performed using the same profile as the first PCR. Ten microliters of each PCR product was analyzed by agarose gel electrophoresis. All positive samples were repeated and sequenced to confirm the presence of HCoV-NL63. To verify negative and positive PCR results, an additional diagnostic RT-PCR assay was conducted using the 1a gene primers 5′-AATATGTCTAACAAATAAAACGATT-3′ (reverse transcriptase primer P4H10-3; coordinate 6667), 5′-CTTTTGATAACGGTCACTATG-3′ (SS 5852-5P; coordinate 5777) and 5′-CTCATTACATAAAACATCAAACGG-3′ (P4G1M-5-3P; coordinate 6616) in the first PCR; and 5′-GGTCACTATGTAGTTTATGATG-3′ (P3E2-5P; coordinate 5788) and 5′-GGATTTTTCATAACCACTTAC-3′ (SS 6375-3P; coordinate 6313) in the nested PCR. Details of sequence analysis are available in Supplementary Methods online.
GenBank accession numbers.
The HCoV-NL63 sequences were deposited in GenBank under accession numbers AY567487–AY567494.
Note: Supplementary information is available on the Nature Medicine website.
References
Stohlman, S.A. & Hinton, D.R. Viral induced demyelination. Brain Pathol. 11, 92–106 (2001).
Jubelt, B. & Berger, J.R. Does viral disease underlie ALS? Lessons from the AIDS pandemic. Neurology 57, 945–946 (2001).
Shingadia, D., Bose, A. & Booy, R. Could a herpesvirus be the cause of Kawasaki disease? Lancet Infect. Dis. 2, 310–313 (2002).
Bachem, C.W. et al. Visualization of differential gene expression using a novel method of RNA fingerprinting based on AFLP: analysis of gene expression during potato tuber development. Plant J. 9, 745–753 (1996).
Holmes, K.V. & Lai, M.M.C. Coronaviridae. in Fields Virology (eds. Fields, B.N. et al.) 1075–1093 (Lippincott-Raven Publishers, Philadelphia, 1996).
Guy, J.S., Breslin, J.J., Breuhaus, B., Vivrette, S. & Smith, L.G. Characterization of a coronavirus isolated from a diarrheic foal. J. Clin. Microbiol. 38, 4523–4526 (2000).
Tyrrell, D.A.J. & Bynoe, M.L. Cultivation of novel type of common-cold virus in organ cultures. Br. Med. J. 1, 1467–1470 (1965).
Hamre, D. & Procknow, J.J. A new virus isolated from the human respiratory tract. Proc. Soc. Exp. Biol. Med. 121, 190–193 (1966).
Almeida, J.D. & Tyrrell, D.A. The morphology of three previously uncharacterized human respiratory viruses that grow in organ culture. J. Gen. Virol. 1, 175–178 (1967).
Thiel, V., Herold, J., Schelle, B. & Siddell, S.G. Infectious RNA transcribed in vitro from a cDNA copy of the human coronavirus genome cloned in vaccinia virus. J. Gen. Virol. 82, 1273–1281 (2001).
McIntosh, K., Dees, J.H., Becker, W.B., Kapikian, A.Z. & Chanock, R.M. Recovery in tracheal organ cultures of novel viruses from patients with respiratory disease. Proc. Natl. Acad. Sci. USA 57, 933–940 (1967).
Hendley, J.O., Fishburne, H.B. & Gwaltney, J.M., Jr. Coronavirus infections in working adults. Eight-year study with 229 E and OC 43. Am Rev. Respir. Dis. 105, 805–811 (1972).
Mounir, S., Labonte, P. & Talbot, P.J. Characterization of the nonstructural and spike proteins of the human respiratory coronavirus OC43: comparison with bovine enteric coronavirus. Adv. Exp. Med. Biol. 342, 61–67 (1993).
Kunkel, F. & Herrler, G. Structural and functional analysis of the surface protein of human coronavirus OC43. Virol. 195, 195–202 (1993).
Bradburne, A.F., Bynoe, M.L. & Tyrrell, D.A. Effects of a “new” human respiratory virus in volunteers. Br. Med. J. 3, 767–769 (1967).
Peiris, J.S. et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361, 1767–1772 (2003).
Lai, M.M. SARS virus: the beginning of the unraveling of a new coronavirus. J. Biomed. Sci. 10, 664–675 (2003).
Drosten, C. et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1967–1976 (2003).
Martina, B.E. et al. Virology: SARS virus infection of cats and ferrets. Nature 425, 915 (2003).
Guan, Y. et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302, 276–278 (2003).
Marra, M.A. et al. The genome sequence of the SARS-associated coronavirus. Science 300, 1399–1404 (2003).
Snijder, E.J. et al. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 331, 991–1004 (2003).
Hamparian, V.V. Rhinoviruses. in Diagnostic Procedures for Viral, Rickettsial and Chlamydial Infection. (eds. Lennette, E.H. & Schmidt, N.J.) 562 (American Public Health Association, Washington DC, 1979).
Rota, P.A. et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300, 1394–1399 (2003).
Chen, L.L., Ou, H.Y., Zhang, R. & Zhang, C.T. ZCURVE_CoV: a new system to recognize protein coding genes in coronavirus genomes, and its applications in analyzing SARS-CoV genomes. Biochem. Biophys. Res. Commun. 307, 382–388 (2003).
Sawicki, S.G. & Sawicki, D.L. Coronaviruses use discontinuous extension for synthesis of subgenome-length negative strands. Adv. Exp. Med. Biol. 380, 499–506 (1995).
van Marle, G. et al. Arterivirus discontinuous mRNA transcription is guided by base pairing between sense and antisense transcription-regulating sequences. Proc. Natl. Acad. Sci. USA 96, 12056–12061 (1999).
Lole, K.S. et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160 (1999).
Mounir, S. & Talbot, P.J. Human coronavirus OC43 RNA 4 lacks two open reading frames located downstream of the S gene of bovine coronavirus. Virol. 192, 355–360 (1993).
Duarte, M. et al. Sequence analysis of the porcine epidemic diarrhea virus genome between the nucleocapsid and spike protein genes reveals a polymorphic ORF. Virol. 198, 466–476 (1994).
Godet, M., Grosclaude, J., Delmas, B. & Laude, H. Major receptor-binding and neutralization determinants are located within the same domain of the transmissible gastroenteritis virus (coronavirus) spike protein. J. Virol. 68, 8008–8016 (1994).
Krempl, C. et al. Characterization of the sialic acid binding activity of transmissible gastroenteritis coronavirus by analysis of haemagglutination-deficient mutants. J. Gen. Virol. 81, 489–496 (2000).
Bonavia, A., Zelus, B.D., Wentworth, D.E., Talbot, P.J. & Holmes, K.V. Identification of a receptor-binding domain of the spike glycoprotein of human coronavirus HCoV-229E. J. Virol. 77, 2530–2538 (2003).
Ksiazek, T.G. et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1953–1966 (2003).
Vaughn, E.M., Halbur, P.G. & Paul, P.S. Sequence comparison of porcine respiratory coronavirus isolates reveals heterogeneity in the S, 3, and 3-1 genes. J. Virol. 69, 3176–3184 (1995).
McIntosh, K. et al. Coronavirus infection in acute lower respiratory tract disease of infants. J. Infect. Dis. 130, 502–507 (1974).
Pene, F. et al. Coronavirus 229E-related pneumonia in immunocompromised patients. Clin. Infect. Dis. 37, 929–932 (2003).
Boivin, G. et al. Human metapneumovirus infections in hospitalized children. Emerg. Infect. Dis. 9, 634–640 (2003).
Myint, S., Johnston, S., Sanderson, G. & Simpson, H. Evaluation of nested polymerase chain methods for the detection of human coronaviruses 229E and OC43. Mol. Cell. Probes 8, 357–364 (1994).
Stephensen, C.B., Casebolt, D.B. & Gangopadhyay, N.N. Phylogenetic analysis of a highly conserved region of the polymerase gene from 11 coronaviruses and development of a consensus polymerase chain reaction assay. Virus Res. 60, 181–189 (1999).
Boom, R. et al. A rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 28, 495–503 (1990).
Acknowledgements
We thank G. Koen and H. van Eijk for the images of the CPE, W. van Est for artwork, W. Pauw for providing patient specimens, and A. de Ronde for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
L.v.d.H. is listed as the inventor on a European and a US patent application, held by Primagen Holding b.v., entitled “Coronavirus nucleic acid, protein, and methods for the generation of vaccine, medicaments and diagnostics.”
Rights and permissions
About this article
Cite this article
van der Hoek, L., Pyrc, K., Jebbink, M. et al. Identification of a new human coronavirus. Nat Med 10, 368–373 (2004). https://doi.org/10.1038/nm1024
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm1024
This article is cited by
-
Recombinant protein embedded liposome on gold nanoparticle based on LSPR method to detect Corona virus
Nano Convergence (2023)
-
Systems pharmacology dissection of pharmacological mechanisms of Xiaochaihu decoction against human coronavirus
BMC Complementary Medicine and Therapies (2023)
-
Coronavirus covid-19 detection by means of explainable deep learning
Scientific Reports (2023)
-
Comparative sequence analysis of SARS nCoV and SARS CoV genomes for variation in structural proteins
Proceedings of the Indian National Science Academy (2023)
-
Bacterial ribosomal RNA detection in cerebrospinal fluid using a viromics approach
Fluids and Barriers of the CNS (2022)
