
Abstract
Rationale
Racemic propranolol (PRO), a β-adrenoceptor antagonist, has been evaluated as a test agent but not as a discriminative stimulus. Its S(−) stereoisomer is thought to subserve the effects of (±)PRO.
Materials and methods
Rats were trained to discriminate S(−)PRO (5 mg/kg) from saline in a two-lever food-reinforced operant conditioning task.
Results
The S(−)PRO stimulus was shown to be centrally mediated, dose-related, time dependent, and stereoselective: S(−)PRO (ED50 = 2.2 mg/kg) was twice as potent as (±)PRO and approximately four times as potent as R(+)PRO. The S(−)PRO stimulus generalized fully to the β-adrenoceptor agent pindolol, the α1-adrenoceptor agonist methoxamine, cocaine, and the serotonergic agents TFMPP and RU 24969; partial generalization occurred to (−)ephedrine and nisoxetine but not to fenfluramine or 5-OMe DMT. The S(−)PRO stimulus was blocked completely (and competitively) when prazosin, an α1-adrenoceptor antagonist, was given in combination with the training dose of S(−)PRO. Moreover, prazosin exerted antagonism of the S(−)PRO-like effect of (±)PRO or R(+)PRO but produced only partial antagonism of the S(−)PRO-like effect of cocaine. In a second study, rats were trained to discriminate 8 mg/kg of cocaine from saline. The cocaine stimulus generalized to S(−)PRO, (±)PRO, and R(+)PRO. Prazosin partially attenuated the stimulus effect of cocaine (8 mg/kg) but completely blocked the cocaine-like effects of (±), S(−), and R(+)PRO.
Conclusions
PRO and cocaine exhibited cross-substitution, but their stimulus effects were antagonized differentially by prazosin. PRO (and its optical isomers) can exert a stimulus effect that is based, at least in part, on increased α1-adrenoceptor activity. PRO might be better characterized as an adrenoceptor partial agonist.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Propranolol (PRO) is a nonselective β1/β2-adrenoceptor antagonist that is used therapeutically for the treatment of certain types of cardiovascular conditions (arrhythmias, angina pectoris, and hypertension) and anxiety (e.g., Simpson 1974; Fitzgerald 1980). In some patients, PRO also can produce CNS side effects such as insomnia and depression; such actions are thought to be associated with certain β-adrenoceptor blockers (e.g., propranolol) that are more lipophilic (i.e., more likely to penetrate the blood–brain barrier) than other β-adrenoceptor blockers that are more hydrophilic (e.g., atenolol; Agon et al. 1991; Frishman et al. 1981; Middlemiss et al. 1981; Neil-Dwyer et al. 1981; Woods and Robinson 1981; Petrie et al. 1982). In radioligand binding studies, PRO displays high affinity for β1- and β2-adrenoceptors (e.g., Fraundorfer et al. 1994; Schotte et al. 1996). It also displays moderate affinity for serotonin 5-HT1A and 5-HT1B receptors but binds with much lower, or no, affinity at 5-HT1D, 5-HT2A, 5-HT2C, 5-HT3, α2-adrenoceptors, histamine H1, and dopamine D1 and D2 receptors (e.g., Kanba and Richelson 1984; Engel et al. 1986; Boyajian and Leslie 1987; Schlicker et al. 1989; Hamon et al. 1990; Toll et al. 1998). Furthermore, the chemical structure of PRO contains an asymmetric center and, thus, PRO exists as a pair of optical isomers: S(−)propranolol (S(−)PRO) and R(+)propranolol (R(+)PRO). The enantiomers bind at β1/β2-adrenoceptors, 5-HT1A, and 5-HT1B receptors in a stereoselective manner with relatively large differences in affinity: S(−)PRO is ∼100 times more potent than R(+)PRO at β-adrenoceptors or 5-HT1B receptors and ∼30-fold more potent at 5-HT1A receptors (e.g., Middlemiss 1984; Hoyer et al. 1985a, b; Engel et al. 1986; Pierson et al. 1989; Tsuchihashi et al. 1990).
In addition to its β-adrenoceptor antagonist effects, PRO has been proposed to function in some instances as a central 5-HT receptor antagonist, releaser of norepinephrine (NE), and/or inhibitor of NE reuptake. For example, PRO can antagonize the (a) hypothermia induced by the purported 5-HT1A/7 agent 8-hydroxy-2-(di-n-propylamino)tertralin (8-OH DPAT) in rats (but not in mice; e.g., Goodwin and Green 1985; To et al. 1995; Toll et al. 1998), (b) suppressant effect of 8-OH DPAT applied microiontophoretically to 5-HT dorsal raphe neurons (Sprouse and Aghajanian 1986), and (c) reciprocal forepaw treading (“serotonin syndrome”) in rats induced by 8-OH DPAT or the 5-HT1A/2A/2C/6/7 agent 5-methoxy-N,N-dimethyltryptamine (5-OMe DMT; e.g., Tricklebank 1985; Tricklebank et al. 1985; Hoyer et al. 1989; Sadzot et al. 1989; To et al. 1995; Glennon et al. 2000). PRO also may interact with adrenoceptors in ways other than, or in addition to, antagonism of β-adrenoceptors. For instance, PRO can raise central α-adrenoceptor (primarily α1-adrenoceptor) activity via the release of NE and/or an inhibition of NE reuptake (e.g., Kelliher and Buckley 1970; Day and Roach 1974; Davies and Reid 1975; Kobinger 1978; Privitera et al. 1979; Tackett et al. 1981; Tuross and Patrick 1986). Interestingly, however, R(+)PRO is nearly as potent or slightly less potent (i.e., 1.5- to ∼3-fold) than S(−)PRO with respect to those indirect agonist actions on adrenoceptors (Daniell et al. 1976; Saelens et al. 1977; Tackett et al. 1981; Tuross and Patrick 1986).
PRO also has been widely employed as a test agent in drug discrimination (DD) studies, a useful procedure to examine the central effect(s) of drugs (e.g., Young and Glennon 1986). For example, PRO [antagonism dose 50% (AD50) = 0.005 mg/kg] can block the discriminative stimulus effect of clenbuterol (0.1 mg/kg), a selective β2-adrenoceptor agonist, in rats (O’Donnell 1997; Ordway et al. 1987). In contrast, the stimulus effect of agents such as ethanol, S(+)amphetamine, S(+)methamphetamine, Δ9-THC, morphine, or yohimbine is not blocked by propranolol, and, thus, β-adrenoceptor involvement in their stimulus action has been discounted (e.g., Schechter 1974; Schechter and Cook 1975; Winter 1975; Colpaert et al. 1977; Jarbe and Ohlin 1977; Munzar and Goldberg 1999). PRO also has been evaluated in DD studies that used training agents that might be mediated primarily via serotonergic receptors. In rats trained to distinguish 8-OH DPAT from vehicle, for example, the stimulus was not antagonized by (and did not generalize to) propranolol (Glennon 1986; Cunningham et al. 1987), a finding that questions the robustness of PRO as a 5-HT (5-HT1A?) receptor antagonist. In comparison, investigators have examined the effects of PRO in animals trained to discriminate the nonselective serotonergic (i.e., moderate to high affinity for 5-HT1A/1B/1D/2A/2B and/or 2C receptors) agents TFMPP [1-(3-trifluoromethylphenyl)piperazine] or RU 24969 [5-methoxy-3-(1,2,3,6-tetrahydro-4-pryridyl)-1H-indole] from saline vehicle (e.g., Peroutka 1986; Schlegel and Peroutka 1986; Boess and Martin 1994; Wainscott et al. 1996; Herrick-Davis et al. 1998). In each instance, stimulus generalization (but not stimulus antagonism) occurred to PRO, and the results suggested that propranolol might behave, at least in part, as an agonist at a 5-HT (5-HT1B?) receptor(s) (Glennon et al. 1988; Gardner 1989). Lastly, PRO does not block the stimulus effect of cocaine but has been reported to enhance, generalize partially, or substitute completely in cocaine-trained animals, suggesting an adrenoceptor component of action in the stimulus effect of cocaine (Ho and McKenna 1978; Colpaert et al. 1979; Kleven and Koek 1997). The latter results also are consistent with findings that PRO dose-dependently enhances other behavioral and neurochemical effects mediated by cocaine (e.g., Goldberg and Gonzalez 1976; Harris et al. 1996).
Taken together, the drug discrimination data above appear to indicate that PRO exerts effects that involve multiple mechanisms: a (primary) β-adrenoceptor antagonist component, enhancement of adrenoceptor activity, and/or an agonist effect at a 5-HT receptor(s). To date, however, propranolol (itself) has not been employed as a training drug. Thus, the major goal of the present study was to determine whether S(−)PRO, the more potent and (presumed) pharmacologically salient enantiomer, could serve as a discriminative stimulus in rats and to evaluate the effects of various agents that alter monoamine neurotransmission. The drugs chosen for tests of stimulus generalization and/or antagonism included the racemic mixture and R(+)PRO isomer of propranolol, cocaine (nonselective increase in monoamine activities through reuptake inhibition), several standard adrenoceptor agents (methoxamine, pindolol, atenolol, nisoxetine, prazosin, and (−)ephedrine), and several “standard 5-HT agents” (fenfluramine, 5-OMe DMT, TFMPP, and RU 24969). It also was of interest to explore more fully the observation that animals trained to discriminate cocaine would generalize to propranolol. Therefore, a second group of rats was trained to discriminate cocaine from saline vehicle, and tests of stimulus generalization were performed to examine the effects of racemic propranolol and its stereoisomers.
Materials and methods
Subjects
The animals were 14 male Sprague–Dawley rats (Charles River Laboratories) that weighed 280–320 g (i.e., 80% food-restricted weights) at the beginning of the study. The animals were divided into two groups and trained to discriminate either S(−)PRO (5.0 mg/kg; n = 8) or cocaine (8.0 mg/kg; n = 6) from saline vehicle. The rats were housed individually, had unlimited access to drinking water, and were maintained at 80% of their free-feeding body weights by partial food restriction. The protocol to conduct these animal studies was approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Discrimination training
Operant conditioning chambers (Coulbourn Instruments, Whitehall, PA, USA, Rat Model E10-10), housed within sound- and light-attenuated outer chambers, were used. Each chamber contained two (response) levers that were mounted at opposite ends of one wall and were separated by a liquid dipper (Model H14-05RE). When activated, the dipper dispensed 0.01 ml of sweetened condensed milk (diluted 2:1 with tap water) for ∼2.5 s as reinforcement; furthermore, a magazine mini-bulb (40 mA at 28 V) was lighted for ∼2.5 s. Rats were trained to lever respond to a variable interval 15-s schedule of reinforcement on both levers. A session (conducted 5 days/week) was started by the activation of the house-light (28 V) in each chamber, and session duration was 15 min. A pre-session injection interval (PSII) of 15 min was employed. The rats were injected intraperitoneally (i.p.) with either drug (i.e., 5.0 mg/kg of S(−)PRO or 8.0 mg/kg of cocaine) or saline on a random schedule, with the constraint that no more than two consecutive sessions with the training drug or vehicle could occur. For half of the rats in each group, responses on the right-side lever were reinforced when drug was given, and responses on the left-side lever were reinforced after saline administration; these conditions were reversed for the remaining rats in each group. On every fifth day of training, discrimination learning was assessed during an initial 2.5-min non-reinforced (extinction) period followed by a 12.5-min training session. Data collected during the extinction periods included percent appropriate responding on the drug (S(−)PRO or cocaine) lever (number of responses on S(−)PRO- or cocaine-designated lever/total number of responses) and total responses (mean responses/minute). Individual animals were not used in the stimulus generalization or antagonism studies until they met a stringent criteria of ≥80% of their responses on the drug-appropriate lever after administration of their assigned training drug and ≤20% of their responses on the same drug-appropriate lever after administration of saline for at least three consecutive weeks.
Generalization and Antagonism tests
During these phases of the study, maintenance of the training drug/saline discriminations was insured by continuation of training sessions on a daily basis (except on a generalization or antagonism test day; see below). On one of the 2 days before a test, approximately half of the animals in each group would receive their training dose of the training drug, and the other half of the rats in each group would receive saline; after a 2.5-min extinction session, training was continued for 12.5 min. Animals not meeting the original criteria (i.e., ≥80% of total responses on the drug-appropriate lever after administration of training drug and ≤20% of total responses on the drug lever after administration of saline) during the extinction session were excluded from the next generalization or antagonism test session. During investigations of stimulus generalization or antagonism, test sessions were interposed among training sessions. During these tests, the rats were allowed 2.5 min of non-reinforced lever responding and were then removed from the operant conditioning chambers. An odd number of training sessions (usually three or five) separated two test sessions.
S(−)Propranolol Stimulus: Generalization studies
The first generalization tests investigated the time-course and dose-response effects of the S(−)PRO stimulus. The time-course study investigated the effects of decreasing or increasing the PSII between the injection of the S(−)PRO training dose and the beginning of a test session. In addition to the standard 15-min delay, the effects of PSIIs of 5, 60, 90, 120, and 150 min were studied. The dose-response study assessed the S(−)PRO-appropriate responding of the rats following the administration of various doses of S(−)PRO (1.25, 2.5, and 5.0 mg/kg). Doses of S(−)PRO were administered in a random sequence with the standard 15-min PSII before behavioral testing. Stimulus generalization tests in the S(−)PRO-trained rats evaluated the following agents: in order, (±)PRO (2.5, 5.0, 7.5, and 10 mg/kg), R(+)PRO (5.0, 10, 15, and 20 mg/kg), cocaine (1.5, 2.5, 3.0, 6.0, 7.0, and 8.0 mg/kg), TFMPP (0.3, 0.5, and 0.75 mg/kg), RU 24969 (0.25, 0.35, and 0.50), 5-OMe DMT (0.5, 0.75, and 1.0 mg/kg), fenfluramine (1.0, 1.5, 2.0, and 3.0 mg/kg), (−)ephedrine (0.3, 1.0, 2.0, 3.0, 3.5, and 4.0 mg/kg), methoxamine (0.3, 0.6, 1.0, 2.0, and 2.5 mg/kg), nisoxetine (5.0, 7.5, 8.5, 9.0, and 10 mg/kg) and pindolol (5.0, 10, 15, and 20 mg/kg). The substitution tests assessed the percent S(−)PRO-like response produced by the administration of the agents. Doses of the compounds were administered in a random sequence, with a 15-min PSII prior to the 2.5-min extinction test period.
S(−)Propranolol Stimulus: Antagonism studies
In the first antagonism tests, rats were administered the α1-adrenoceptor antagonist prazosin (0.1, 0.3, 1.0, 2.0, 3.0, and/or 4.0 mg/kg) 30 min prior to an injection of the training dose of S(−)PRO (5.0 mg/kg), 10 mg/kg of (±)PRO, 20 mg/kg of R(+)PRO, or 8.0 mg/kg of cocaine; each dose of the latter three agents produced a S(−)PRO-like response that met criteria for complete stimulus substitution. A subsequent 15 min elapsed before the animals were exposed to the extinction test session as described above. An initial dose of 0.1 mg/kg of prazosin was used. A second antagonism test evaluated the effect of the β1-adrenoceptor antagonist atenolol (3.0, 10, and 20 mg/kg) on the S(−)PRO stimulus. Doses of atenolol were administered 30 min prior to an injection of the 5 mg/kg training dose of S(−)PRO. An initial dose of 3.0 mg/kg of atenolol was used. The degree of antagonism produced by initial doses of each agent was used to determine subsequent doses of the antagonists. Moreover, control tests of prazosin (0.1, 0.3, 1.0, 2.0, and 3.0 mg/kg) and atenolol (1.0, 3.0, 10, 15, 20, and 30 mg/kg), given alone, were conducted to evaluate their potential to produce a S(−)PRO-like response; a 45-min PSII was used to evaluate each agent. In a third set of antagonism tests, dose-response functions of S(−)PRO were determined in the presence of 1.0 mg/kg or 2.0 mg/kg of prazosin. In these tests, prazosin was administered 30 min before an injection of S(−)PRO, which was followed by an elapsed time of 15 min. Eight rats were used in each antagonism test (including control tests of antagonists).
Cocaine Stimulus: Generalization studies
Stimulus generalization tests assessed the rats’ percentage of cocaine-appropriate responding following administration of various doses of cocaine. Doses of cocaine were administered in a random sequence with the standard 15-min PSII before behavioral testing. Further stimulus generalization tests in these rats evaluated the following agents: in order, S(−)PRO (2.5, 5.0, 10, 12.5, 15, and 20 mg/kg), (±)PRO (12.5, 15, and 20 mg/kg), and R(+)PRO (10, 15, 20, and 25 mg/kg). Substitution tests with these agents assessed the percent cocaine-like response produced by the administration of the agents. Doses of the compounds were administered in a random sequence with a 15-min PSII prior to 2.5-min extinction test periods.
Cocaine Stimulus: Antagonism studies
In antagonism tests, rats were administered the α1-adrenergic receptor antagonist prazosin (0.3, 1.0, 3.0, 4.0, and/or 4.5 mg/kg) 30 min prior to an injection of the training dose of cocaine (8.0 mg/kg), 15 mg/kg of S(−)PRO, 20 mg/kg of (±)PRO, or 25 mg/kg of R(+)PRO; each dose of the latter three agents produced a cocaine-like response that met criteria for complete stimulus substitution. A subsequent 15-min PSII elapsed before the animals were exposed to the extinction test session as described above. An initial dose of 0.3 mg/kg of prazosin was used. In addition, control tests of prazosin (0.3, 0.6, 1.0, 2.0, 3.0, 4.0, and 5.0 mg/kg), given alone, were conducted to evaluate its potential to produce a cocaine-like response; a 45-min PSII was used to evaluate the antagonist. Six rats were used in each antagonism test (including control tests of prazosin).
Data analysis
A determination of complete, partial, or no generalization (or antagonism) was based on previously described criteria (e.g., Khorana et al. 2008; Young and Glennon 1986). In this scheme, the results of stimulus generalization (or antagonism) tests are characterized as one of three possible results: (a) complete stimulus generalization resulted when the animals, following a given dose of drug or drug combination, made ≥80% (i.e., group mean) of their responses on the drug-appropriate lever; (b) no generalization (i.e., saline-like responding) occurred when the test agent produced 0–20% drug-appropriate responding; and (c) partial generalization occurred when a test drug produced an intermediate level of responding (i.e., 21% to 79% but usually between 40% and 70%) on the drug-appropriate lever. Animals that made fewer than five total responses during the 2.5-min extinction session were characterized as being disrupted because they failed to meet the testing criteria. Thus, percent drug-appropriate responding and response rate data refer only to animals making ≥5 responses during the extinction session (Young and Glennon 1986). Data for a particular drug dose were not plotted; however, if >50% of the animals were behaviorally disrupted following administration of that dose. Where complete stimulus generalization (or antagonism) occurred, potency comparisons were made between the training drug and the test agent via calculation of the effective dose 50% (ED50) or antagonism dose 50% (AD50) as determined from the dose-response data by the method of Finney (1952). The ED50 or AD50 value represents the calculated drug dose where animals would be expected to make 50% of their responses on the drug-appropriate lever. The response rate data (i.e., responses/min) of each training group were evaluated by repeated measures ANOVA and followed, when appropriate, by Dunnett’s t test (statistical significance set at p < 0.05) for comparison of a control group (i.e., mean response rate after saline) versus doses(s) of training drug, experimental dose groups, or dose combinations. In the S(−)PRO versus saline group, statistical evaluation of response rate data was found to be significant (F (22, 720) = 8.76, p < 0.01), whereas in the cocaine versus saline group, it was not (F (9, 249) = 0.85, p > 0.05).
Drugs
Cocaine HCl, S(−)propranolol HCl, (±)propranolol HCl, R(+)propranolol HCl, methoxamine HCl, prazosin HCl, (−)ephedrine HCl, atenolol, and pindolol were purchased from Sigma Chemical Company (St. Louis, MO, USA). Fenfluramine HCl, nisoxetine HCl, and 5-methoxy-3-(1,2,3,6-tetrahydro-4-pryridyl)-1H-indole (RU 24969) were gifts from A.H. Robins Company (Richmond, VA, USA), Eli Lilly and Company (Indianapolis, IN, USA), and Roussell-Uclatf (Romainville, France), respectively. 5-Methoxy-N,N-dimethyltryptamine hydrogen oxalate (5-OMe DMT) was synthesized in our laboratory, and TFMPP (1-(3-trifluoromethylphenyl)piperazine HCl) was obtained from Research Biochemicals (Natick, MA, USA). Pindolol and atenolol (free bases) were first dissolved in one equivalent of 0.01 N hydrochloric acid before dilution with saline. The other drugs were dissolved in 0.9% sterile saline, and doses of each compound were based on the weight of the stated salt or base. Drugs were administered i.p. in a 1-ml/kg injection volume, except for doses ≥10 mg/kg of (±), S(−), or R(+)propranolol, which were administered in a 2-ml/kg injection volume.
Results
Figure 1 depicts the rats’ learning of the S(−)PRO (5.0 mg/kg) versus saline vehicle discrimination; approximately 55 training sessions [range = 30 to 40 sessions to (initially) meet criteria] were required to reach criteria. Once achieved, five additional training sessions were conducted to ensure stable performance. Thus, after 60 training sessions, the rats’ discrimination performance was stable (i.e., S(−)PRO, ∼90%; saline, <10%); response rates were similar under the treatment conditions.

Rats acquisition of discriminative control of responding by 5.0 mg/kg of S(−)propranolol (S(−)PRO) versus saline vehicle (1.0 ml/kg of 0.9% saline). Ordinate Mean (±SEM; n = 8) percentage of responses made on the S(−)PRO-appropriate lever after the i.p. administration of S(−)PRO (solid squares) or saline (open squares). Abscissa Test sessions plotted on a linear scale
Rats’ learning of cocaine (8.0 mg/kg) versus saline discrimination has been described previously (Khorana et al. 2004; Young and Glennon 1997).
The time course of stimulus effect produced by 5.0 mg/kg of S(−)PRO is shown in Fig. 2. When the PSII was shortened to 5 min, the rats responded 40% on the S(−)PRO-designated lever. After a PSII of 60 min, S(−)PRO-appropriate responding (91%) was similar to that observed following the standard PSII of 15 min. PSIIs subsequent to 60 min resulted in decreased percentages of total responses on the S(−)PRO-appropriate lever; with PSIIs of 120 and 150 min, drug-designated lever responding was lowered to 39% and 11%, respectively. The rats’ response rates were not statistically different (Dunnett’s t; p > 0.05) from saline treatment(s) in the time-course study of S(−)PRO (Fig. 2, lower graph).

Time course of the S(−)PRO (5.0 mg/kg) discriminative stimulus in rats (n = 8) trained to discriminate 5.0 mg/kg of S(−)PRO from saline vehicle. Ordinate Mean (±SEM) percentage of responses made on the S(−)PRO-appropriate lever after the i.p. administration of 5.0 mg/kg of S(−)PRO followed by various pre-session injection intervals (top panel). The animals’ response rates are shown in the lower panel. Abscissa Pre-session injection interval (minutes) plotted on a linear scale
S(−)Propranolol Stimulus: generalization studies
As shown in Fig. 3, the S(−)PRO (ED50 = 2.2 mg/kg; 95% CL = 1.3–3.6 mg/kg) stimulus generalized to (±)PRO (ED50 = 4.3 mg/kg; 95% CL = 3.0–6.4 mg/kg) and R(+)PRO (ED50 = 8.4 mg/kg; 95% CL = 5.8–12.2 mg/kg). A comparison of ED50 values revealed that S(−)PRO is approximately twice as potent as the racemic mixture and approximately four times more potent than R(+)PRO. Figure 4 shows that the S(−)PRO stimulus generalized to the following agents: TFMPP (ED50 = 0.5 mg/kg; 95% CL = 0.32–0.74 mg/kg), RU 24969 (ED50 = 0.35 mg/kg; 95% CL = 0.28–0.43 mg/kg), methoxamine (ED50 = 1.2 mg/kg; 95% CL = 0.65–2.1 mg/kg), pindolol (ED50 = 9.0 mg/kg; 95% CL = 5.6–14.4 mg/kg), and cocaine (ED50 = 2.3 mg/kg; 95% CL = 1.3–3.8 mg/kg); eight out of eight rats responded/tested at each dose except six out of eight at 2.0 or 2.5 mg/kg of methoxamine. Figures 3 and 4 also indicate that the animals’ response rates were not appreciably altered, in comparison to that after administration of saline, following injections of S(−)PRO, (±)PRO, R(+)PRO, pindolol, TFMPP, or RU 24969, but were significantly reduced (by 50 to 75%) after administration of methoxamine (2.5 mg/kg) or cocaine (8.0 mg/kg).

S(−)PRO stimulus generalization to (±), S(−), and R(+)PRO in rats (n = 8) trained to discriminate 5.0 mg/kg of S(−)PRO from saline vehicle. Ordinate Mean (±SEM) percentage of responses made on the S(−)PRO-appropriate lever after the i.p. administration of various doses of each drug (top figure). The animals’ response rates are shown in the lower figure. D Responses following 5.0 mg/kg of S(−)PRO and S responses following administration of saline vehicle. Abscissa Drug doses plotted on a logarithmic scale

S(−)PRO stimulus generalization to cocaine, methoxamine, pindolol, TFMPP, and RU 24969 in rats (n = 8) trained to discriminate 5.0 mg/kg of S(−)propranolol from saline vehicle. Ordinate Mean (±SEM) percentage of responses made on the S(−)PRO-appropriate lever after the i.p. administration of various doses of each drug (top figure). The animals’ response rates are shown in the lower figure. D Responses following 5.0 mg/kg of S(−)PRO and S responses following administration of saline vehicle. Response rate was significantly (*p < 0.05) different from the saline vehicle control response rate. Abscissa Drug doses plotted on a logarithmic scale
Figure 5 depicts S(−)PRO partial stimulus generalization to (−)ephedrine (maximum 56% S(−)PRO-appropriate responding at 3.5 mg/kg by five out of eight rats that responded) and disruption of behavior (i.e., zero out of eight rats responded) at 4.0 mg/kg and nisoxetine (maximum 68% S(−)PRO-appropriate responding at 8.5 mg/kg by eight out of eight rats responded) and disruption of behavior (i.e., <50% of rats responded) at 9.0 mg/kg (three out of eight rats responded) or 10 mg/kg (one out of eight rats responded). In addition, the animals’ response rates were significantly reduced following injection of nisoxetine (8.5 mg/kg) or (−)ephedrine (3.0 mg/kg and 3.5 mg/kg). The S(−)PRO stimulus did not generalize to the other drugs: 5-OMeDMT and fenfluramine produced a maximum of 18% and 30% S(−)PRO-appropriate responding at 0.75 mg/kg (five out of eight rats responded) and 1.5 mg/kg (eight out of eight rats responded), respectively; a higher dose(s) of each agent resulted in disruption of behavior (<50% of rats responded), i.e., two out of eight rats responded at 1.0 mg/kg of 5-OMeDMT or 2.0 mg/kg of fenfluramine and, one out of eight rats responded at 3.0 mg/kg of fenfluramine.

Results of stimulus generalization tests conducted with (−)ephedrine, nisoxetine, fenfluramine, and 5-OMeDMT in rats (n = 8) trained to discriminate 5.0 mg/kg of S(−)PRO from saline vehicle. Ordinate Mean (±SEM) percentage of responses made on the S(−)PRO-appropriate lever after the i.p. administration of various doses of each drug (top figure). The animals’ response rates are shown in the lower figure. D Responses following 5.0 mg/kg of S(−)PRO and S responses following administration of saline vehicle. Response rate was significantly (*p < 0.05) different from the saline vehicle control response rate. Abscissa Drug doses plotted on a logarithmic scale. See “Results” for further descriptions of data
S(−)Propranolol Stimulus: antagonism studies
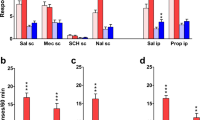
Control tests of prazosin (0.1, 0.3, 1.0, 2.0, and 3.0 mg/kg) or atenolol (1.0, 3.0, 10, 15, 20, and 30 mg/kg), given alone, indicated that neither antagonist produced a S(−)PRO-like response (Fig. 6); seven out of eight rats responded at 3.0 mg/kg of prazosin; four and one out of eight rats responded at 20.0 and 30.0 mg/kg of atenolol, respectively. It also was noted that the animals’ response rates were significantly reduced (20 mg/kg) or completely disrupted (30 mg/kg) following injection of atenolol. Figure 6 also shows that the administration of prazosin prior to injection of the (a) training dose of S(−)PRO (i.e., 5.0 mg/kg) or (b) dose of (±)PRO (i.e., 10 mg/kg) or R(+)PRO (i.e., 20 mg/kg) that produced complete stimulus generalization resulted in dose-related antagonism of the S(−)PRO or S(−)PRO-like response: Eight out of eight rats responded/tested at each dose combination. A comparison of AD50 values revealed that prazosin displayed approximately equal potency to block the three agents: S(−)PRO (prazosin AD50 = 0.47 mg/kg; 95% CL = 0.17–0.82 mg/kg), (±)PRO (prazosin AD50 = 0.49 mg/kg; 95% CL = 0.17–0.82 mg/kg), or R(+)PRO (prazosin AD50 = 0.71 mg/kg; 95% CL = 0.17–2.1 mg/kg). Figure 6 also shows that the animals’ response rates were not appreciably altered, in comparison to that after administration of saline, following injections of doses of prazosin in combination with the test dose of S(−), (±), or R(+)PRO. In comparison, prazosin produced only partial stimulus antagonism of the S(−)PRO-like response exerted by 8.0 mg/kg of cocaine (the maximum decrease was to only 63% S(−)PRO-appropriate responding at 3.0 mg/kg of prazosin and 88% S(−)PRO-appropriate responding at 4.0 mg/kg of prazosin): Eight out of eight rats responded at each dose combination. The animals’ response rates, however, were significantly reduced following injection of prazosin (3.0 or 4.0 mg/kg) in combination with cocaine (8.0 mg/kg). In contrast, the administration of atenolol prior to injection of the training dose of S(−)PRO (i.e., 5.0 mg/kg) produced no antagonism of the S(−)PRO stimulus effect (Fig. 6). The combination of 3.0 or 10 mg/kg of atenolol and 5.0 mg/kg of S(−)PRO produced 95% and 98% S(−)PRO-appropriate responding, respectively; a higher dose (i.e., 20 mg/kg) of atenolol resulted in disruption of behavior (i.e., zero out of eight rats responded).

Results of stimulus antagonism tests with various doses of prazosin or atenolol given alone or in combination with a dose of S(−)PRO (5.0 mg/kg), (±)PRO (10 mg/kg), R(+)PRO (20 mg/kg), or cocaine (8 mg/kg) in rats (n = 8) trained to discriminate 5.0 mg/kg of S(−)propranolol from saline vehicle. Ordinate Mean (±SEM) percentage of responses (top figure) made on the S(−)PRO-appropriate lever after the i.p. administration of the various doses of each antagonist given before the dose of each test drug. The animals’ response rates are shown in the lower figure. D Responses following 5.0 mg/kg of S(−)PRO and S responses following administration of saline vehicle. Response rate was significantly (*p < 0.05) different from the saline vehicle control response rate. Abscissa Drug doses plotted on a logarithmic scale. See “Results” for further descriptions of data
Lastly, Fig. 7 shows that pretreatment of the S(−)PRO-trained animals with 1.0 or 2.0 mg/kg of prazosin in combination with various doses of S(−)PRO resulted in parallel shifts of the dose-response functions and, consequently, changes in the ED50 dose of S(−)PRO: S(−)PRO given alone (ED50 = 2.2 mg/kg; 95% CL = 1.3–3.6 mg/kg), 1.0 mg/kg of prazosin and doses of S(−)PRO (ED50 = 5.4 mg/kg; 95% CL = 2.8–10.5 mg/kg), 2.0 mg/kg of prazosin and doses of S(−)PRO (ED50 = 10.4 mg/kg; 95% CL = 6.0–18.0 mg/kg). In the latter tests, the animals’ response rates were not markedly different, in comparison to that after administration of saline, following injections of those doses of prazosin in combination with doses of S(−)PRO (Dunnett’s t; p > 0.05).

Results of stimulus antagonism tests with various doses of S(−)PRO given alone or in combination with doses of prazosin (1.0 or 2.0 mg/kg) in rats (n = 8) trained to discriminate 5.0 mg/kg of S(−)propranolol from saline vehicle. Ordinate Mean (±SEM) percentage of responses (top figure) made on the S(−)PRO-appropriate lever after the i.p. administration of the various doses of each antagonist before the dose of S(−)PRO. The animals’ response rates are shown in the lower figure. D Responses following 5.0 mg/kg of S(−)PRO and S responses following administration of saline vehicle. Abscissa Drug doses plotted on a logarithmic scale
Cocaine Stimulus: generalization studies
As shown in Fig. 8, the cocaine (ED50 = 2.7 mg/kg; 95% CL = 1.5–4.7 mg/kg) stimulus generalized to S(−)PRO (ED50 = 12.2 mg/kg; 95% CL = 10.4–14.2 mg/kg), (±)PRO (ED50 = 15.9 mg/kg; 95% CL = 12.6−20.1 mg/kg), and R(+)PRO (ED50 = 17.8 mg/kg; CL = 13.7–23.1 mg/kg). A comparison of ED50 values revealed that the cocaine-like response of S(−)PRO was <1.5 times more potent than the racemic mixture and 1.5 times more potent than the R(+)PRO enantiomer. The animals’ response rates were not appreciably altered, in comparison to that after administration of saline, following injections of cocaine, S(−), (±), or R(+)PRO.

Cocaine-stimulus generalization to cocaine, (±), S(−), and R(+)PRO in rats (n = 6) trained to discriminate 8.0 mg/kg of cocaine from saline vehicle. Ordinate Mean (±SEM) percentage of responses made on the cocaine-appropriate lever after the i.p. administration of various doses of each drug (top figure). The animals’ response rates are shown in the lower figure. D Responses following 8.0 mg/kg of cocaine and S responses following administration of saline vehicle. Abscissa Drug doses plotted on a logarithmic scale
Cocaine Stimulus: antagonism studies
Control tests of prazosin (0.3, 0.6, 1.0, 2.0, 3.0, 4.0, and 5.0 mg/kg), given alone, indicated that it did not produce a cocaine-like effect (Fig. 9); five out of six rats responded at 2.0, 3.0, and 5.0 mg/kg of prazosin. Furthermore, the animals’ response rates were not markedly changed, in comparison to that after administration of saline, following injection of doses of prazosin. Figure 9 also indicates that the administration of prazosin prior to the injection of S(−)PRO (15 mg/kg), (±)PRO (20 mg/kg), or R(+)PRO (25 mg/kg), doses that produced complete stimulus generalization in the cocaine-trained animals, resulted in a dose-related antagonism of each cocaine-like response: Six out of six rats responded/tested at each dose combination. A comparison of AD50 values revealed that prazosin displayed nearly equal potency to block the three agents: S(−)PRO (prazosin AD50 = 1.7 mg/kg; 95% CL = 0.8–3.7 mg/kg), (±)PRO (prazosin AD50 = 1.5 mg/kg; 95% CL = 0.7–3.1 mg/kg), or R(+)PRO (prazosin AD50 = 1.2 mg/kg; 95% CL = 0.5–2.7 mg/kg). The animals’ response rates were not appreciably altered, in comparison to that after administration of saline, following injections of doses of prazosin in combination with the test dose of S(−), (±), or R(+)PRO. In comparison, prazosin produced only partial stimulus antagonism of the stimulus produced by 8.0 mg/kg of cocaine (the maximum decrease was to only 64% cocaine-appropriate responding at 4.0 mg/kg of prazosin and 89% cocaine-appropriate responding at 5.0 mg/kg of prazosin); four and five out of six rats responded at the combination of 3.0 or 5 mg/kg of prazosin and cocaine, respectively. In addition, the animals’ response rates were not changed appreciably following injection of prazosin (0.3 to 5.0 mg/kg) in combination with 8.0 mg/kg of cocaine.

Results of stimulus antagonism tests with various doses of prazosin given alone or in combination with a dose of cocaine (8.0 mg/kg), S(−)PRO (15 mg/kg), (±)PRO (20 mg/kg), or R(+)PRO (25 mg/kg) in rats (n = 6) trained to discriminate 8.0 mg/kg of cocaine from saline vehicle (1 ml/kg). Ordinate Mean (±SEM) percentage of responses (top figure) made on the cocaine-appropriate lever after the i.p. administration of the various doses of prazosin before the dose of each test drug. The animals’ response rates are shown in the lower figure. D Responses following 8.0 mg/kg of cocaine and S responses following administration of saline vehicle. Abscissa Drug doses plotted on a logarithmic scale. See “Results” for further descriptions of data
Discussion
This study demonstrates quite clearly that S(−)PRO, at 5.0 mg/kg, can serve as a discriminative stimulus in rats. The time-course investigation of the S(−)PRO stimulus revealed a relatively fast onset to action and a relatively short duration of effect. Indeed, the latter results parallel pharmacokinetic studies of propranolol in rats that indicate PRO is rapidly and completely absorbed after intraperitoneal administration and that its peak plasma level is observed within 1 h and steadily declines thereafter (Fitzgerald 1980; Hayes and Cooper 1971). In addition, S(−)PRO- or cocaine-stimulus generalization tests indicated that S(−)PRO is <1.5 to two times more potent than (±)PRO and 1.5 to four times more potent than R(+)PRO. Thus, the discriminative stimulus effect(s) of PRO appears to reside primarily, albeit not exclusively, with the S(−)-isomer. More importantly, however, both groups of animals produced potency relationships between S(−), (±), and R(+)PRO that strongly suggest, but do not conclusively prove, that the stimulus effect of PRO (at the doses tested) is unlikely to be mediated through β-adrenoceptors. Specifically, the S(−)PRO- and cocaine-like stimulus effectiveness of R(+)PRO, which is approximately 100 times less potent than S(−)PRO in blocking cardiac β-adrenoceptors and in radioligand β-adrenoceptor binding studies in brain (Barrett and Cullum 1968; Tsuchihashi et al. 1990), is similar to that of the S(−)-isomer. The relatively slight difference in potency between the stereoisomers is much more consistent with their potency to release NE or inhibit NE reuptake and, consequently, increase adrenoceptor activity (Daniell et al. 1976; Saelens et al. 1977; Tackett et al. 1981; Tuross and Patrick 1986; also see Adrenergic involvement below).
Serotonergic involvement
The S(−)PRO stimulus did not generalize to the nonselective 5-HT agents 5-OMe DMT or fenfluramine. 5-OMe DMT has been shown to bind to 5-HT1A/1B/2A/2C/6 and 7 receptors (Glennon et al. 2000; Gozlan et al. 1983; Lyon and Titeler 1988; Offord et al. 1988; Peroutka 1986; Sadzot et al. 1989; To et al. 1995). Fenfluramine is known to release endogenous stores of 5-HT, an effect that should allow released 5-HT to interact at 5-HT receptors in a nonselective manner. If the S(−)PRO stimulus is mediated to some extent by 5-HT receptors, then it might be anticipated that 5-OMe DMT and fenfluramine would exert some degree of partial, if not complete, stimulus generalization. Such was not the case because both drugs produced saline-like responding at the highest non-disruptive doses tested. Thus, the results suggest a negligible contribution of 5-HT in the stimulus effect of S(−)PRO. However, it should be noted that 5-OMe DMT and fenfluramine have sometimes produced stimulus results that could be termed inconsistent when compared with other “serotonergically mediated” agents. For example, in 5-OMe DMT-trained animals, stimulus generalization does not occur to fenfluramine but does occur to 8-OH DPAT (Spencer et al. 1987). In contrast, fenfluramine-trained animals generalize to 5-OMe DMT but not to the 5-HT1A receptor agonist 8-OH DPAT (Young and Glennon 1988). Lastly, an 8-OH DPAT stimulus does not generalize to fenfluramine (Glennon 1986) and, reportedly, does not generalize (Cunningham and Appel 1986; Glennon 1986) or only partially generalizes to 5-OMe DMT (Tricklebank et al. 1987).
In contrast to the results obtained with 5-OMe DMT and fenfluramine, S(−)PRO stimulus generalization occurred to TFMPP and RU 24969. Early studies of the latter two agents as discriminative stimuli emphasized the possible role of 5-HT1B receptors in the mediation of their effects. That conclusion was based on the absence of stimulus generalization to compounds “selective” for other subpopulations of 5-HT receptors as well as upon generalization to other purported “5-HT1B receptor drugs”. For example, in animals trained to discriminate either TFMPP or RU 24969 from vehicle, stimulus generalization occurred to the purported “5-HT1B agents” 1-(m-chlorophenyl)piperazine, TFMPP, and RU 24969, but not to 8-OH DPAT (Cunningham and Appel 1986; Gardner 1989; Glennon et al. 1984; McKenney and Glennon 1986). Furthermore, both a TFMPP and a RU 24969 stimulus generalized to PRO, which seemed to suggest an agonist effect of PRO at 5-HT1B receptors (Gardner 1989; Glennon et al. 1988). Later studies with TFMPP and RU 24969, however, determined that these agents were nonselective 5-HT agonists. For example, TFMPP and RU 24969 are now known to exhibit moderate to high affinity for 5-HT1A/1B/1D/2A/2B and/or 2C receptors (e.g., Peroutka 1986; Schlegel and Peroutka 1986; Boess and Martin 1994; Wainscott et al. 1996; Herrick-Davis et al. 1998). Lastly, it has been reported that a TFMPP stimulus is not blocked by the α1-adrenoceptor antagonist prazosin but, curiously, is blocked by the nonselective β-adrenoceptor antagonist (−)-alprenolol (Arnt 1989; Herndon et al. 1992). Nevertheless, even though the present results with 5-OMe DMT and fenfluramine might not suggest a role for 5-HT in the stimulus effect of S(−)PRO, the current findings with TFMPP and RU 24969 still hint that a role for 5-HT cannot be assumed to have been definitively eliminated as a factor in the stimulus mechanism of action of PRO.
Investigations with cocaine
The S(−)PRO stimulus generalized to cocaine, and the cocaine stimulus generalized to racemic propranolol and its enantiomers with the following order of potency: S(−)PRO > (±)PRO > R(+)PRO. Cocaine interacts with multiple neurotransmitter systems such that it blocks neuronal reuptake of the monoamines dopamine, norepinephrine, and serotonin (e.g., Koe 1976; Rothman et al. 2001). Drug discrimination studies also provide several lines of evidence that indicate the stimulus actions of cocaine are mediated, at least in part, through modulatory actions by dopamine, 5-HT, and NE systems (e.g., Cunningham and Callahan 1991; Kleven and Koek 1997; Terry et al. 1994). In the present tests of stimulus antagonism in the cocaine- or S(−)PRO-trained animals, the administration of prazosin before cocaine produced only partial antagonism. In comparison, the administration of prazosin prior to racemic propranolol (or its stereoisomers) in the cocaine-trained rats produced dose-related antagonism of cocaine-like appropriate responding; prazosin is almost equipotent as an antagonist of the cocaine-like effect of each agent. The latter results are in agreement and consistent with a previous report that prazosin produced partial antagonism of a cocaine stimulus and that prazosin antagonized fully the cocaine-enhancing stimulus effects of PRO in rats (Kleven and Koek 1998). On the other hand, prazosin has been reported to produce either no antagonism (at all) of the stimulus effect of cocaine in rats (Kleven et al. 1999) or full antagonism (or a shift of the dose response) of the stimulus effect of cocaine in pigeons (Johanson and Barrett (1993) and squirrel monkeys (Rowlett et al. 2004; Spealman 1995). Taken together, the latter results raise the likely possibility of a species (and/or other methodological) difference(s) in the behavioral evaluation of the interactive effects between prazosin and cocaine. Nonetheless, the current data obtained in the cocaine- and S(−)PRO-trained animals strongly re-emphasize the idea that stimulus generalization between a training drug and a test drug is only evidence that both agents can produce a similar stimulus effect and that it is not necessarily accurate to conclude that they do so via an identical mechanism of action (e.g., Young and Glennon 1986).
Adrenergic involvement
In this initial study, S(−)PRO stimulus generalization and antagonism tests were conducted to gauge the degree of involvement of adrenoceptors. The S(−)PRO stimulus was not influenced (in substitution or antagonism tests) by atenolol. The latter results could be due to the hydrophilic character of atenolol, an attribute that should restrict its entry into the brain as compared to the highly lipophilic nature of PRO (e.g., Agon et al. 1991). Moreover, the results with atenolol promote the idea of a (primary) central site of stimulus action for S(−)PRO. It should be noted, however, that atenolol is a relatively selective peripheral β1-adrenoceptor agent and, therefore, it would not account for the possible involvement of peripheral β2-adrenoceptors in the stimulus effect of S(−)PRO. In other tests of stimulus substitution, the S(−)PRO stimulus generalized partially to the selective NE reuptake inhibitor nisoxetine and the nonselective adrenoceptor agonist (−)ephedrine. In as much as S(−)PRO stimulus generalization might have occurred at higher doses had animals’ behavior not been disrupted, the possibility exists that those partial (although incomplete) generalizations may indicate an increase in adrenergic activity as a component of the stimulus effect of S(−)PRO. That notion was confirmed by determinations of S(−)PRO stimulus generalization to pindolol, a nonselective β-adrenoceptor antagonist that also (at relatively high doses) exerts intrinsic sympathomimetic (agonist) activity (ISA; e.g., Frishman 1988; Gonasun and Langrall 1982), and methoxamine, an α1-adrenoceptor agonist. Indeed, a series of antagonism tests appear to confirm that the S(−)PRO stimulus is comprised of an α1-adrenoceptor agonist effect. First, the administration of prazosin prior to the injection of (±)PRO or its stereoisomers produced dose-related antagonism of S(−)PRO (or S(−)PRO-like) appropriate responding; prazosin is almost equipotent as an antagonist of their effects. The S(−)PRO-trained animals also were subjected to a more extensive series of antagonism tests with pretreatment of 1 or 2 mg/kg of prazosin prior to the administration of various doses of S(−)PRO. Those treatments produced (parallel) rightward shifts of the dose-response curves and, consequently, marked changes in the ED50 dose of S(−)PRO; ED50 values of S(−)PRO shifted 2.5- and five-fold when S(−)PRO was given alone versus when it was given in the presence of 1 and 2 mg/kg of prazosin, respectively. Thus, it is possible that PRO and its stereoisomers might exert a central effect(s) that arises from an increased release or a decreased reuptake of NE into central adrenergic nerve terminals (e.g., Kelliher and Buckley 1970; Day and Roach 1974; Davies and Reid 1975; Kobinger 1978; Privitera et al. 1979; Tackett et al. 1981; Tuross and Patrick 1986). However, the present experiments cannot discern inhibition of reuptake and/or stimulation of release of NE by PRO, but in either, or both, case(s), the result would be an increase in adrenergic action in the synapse. That prior treatment with prazosin prevents the S(−)PRO stimulus effect suggests that the action may be mediated (primarily) through stimulation of central α1-adrenoceptors. In fact, the data demonstrate the competitive relationship between the seemingly α1-adrenoceptor agonist effect of S(−)PRO and the α1-adrenoceptor antagonist effect of prazosin.
Summary
The present study provides evidence that S(−)PRO can serve as a discriminative stimulus in rats. The S(−)PRO stimulus appears to be mediated centrally and is dose-related, time dependent, and stereoselective. The potency relationships between the stimulus effects of (±)PRO and its enantiomers emphasize the importance of steric factors and also suggest an action(s) of PRO in the CNS that may not depend upon β-adrenoceptor antagonism. The S(−)PRO stimulus generalized to the nonselective 5-HT agents TFMPP and RU 24969, which suggests the possibility, to some degree, of a serotonergic component in its mechanism of action. The S(−)PRO stimulus also generalized to the nonselective β-adrenoceptor partial agonist pindolol and the α1-adrenoceptor agonist methoxamine. Consistent with the proposal of PRO-induced activation of α1-adrenoceptors are the findings that the administration of the α1-adrenoceptor antagonist prazosin can block the stimulus effect of S(−)PRO (completely and competitively) and the S(−)PRO-like effect of (±)PRO and R(+)PRO. Thus, even though PRO (at low doses) has been demonstrated to block a β-adrenoceptor agonist stimulus (i.e., clenbuterol), the present study of PRO (at a higher dose) implicates an increase in α1-adrenoceptor activity. Finally, S(−)PRO and cocaine display cross generalization regardless of which agent is used as the training stimulus, but the α1-adrenoceptor antagonist effect of prazosin can reveal differences in their mechanisms of action. The latter results re-confirm the concept that stimulus generalization between a training drug and a test compound is simply evidence that both agents can produce a similar stimulus effect but it is not necessarily accurate to conclude that they do so through an identical mechanism of action. Overall, the present results provide support for the idea that PRO can increase central α1-adrenoceptor activity. Future studies of PRO should consider its dose-dependent effects and not necessarily assume that every dose of racemic PRO or its stereoisomers will exert characteristics of a β-adrenoceptor antagonist effect. In fact, PRO might be described more appropriately as a partial adrenoceptor agonist.
References
Agon P, Goethals P, Van Haver D, Kaufman JM (1991) Permeability of the blood–brain barrier for atenolol studied by positron emission tomography. J Pharm Pharmacol 43:597–600
Arnt J (1989) Characterization of the discriminative stimulus properties induced by 5-HT1 and 5-HT2 agonists in rats. Pharmacol Toxicol 64:165–172
Barrett AM, Cullum VA (1968) The biological properties of the optical isomers of propranolol and their effects on cardiac arrhytmias. Br J Pharmacol 34:43–55
Boess FG, Martin IL (1994) Molecular biology of 5-HT receptors. Neuropharmacology 33:275–317
Boyajian CL, Leslie FM (1987) Pharmacological evidence for α2-adrenoceptor heterogeneity: differential binding properties of [3H]rauwolscine and [3H]idazoxan in rat brain. J Pharmacol Exp Ther 241:1092–1098
Colpaert FC, Niemegeers CJE, Janssen PAJ (1977) Catecholamine involvement in the narcotic cue. Arch Int Pharmacodyn 226:339–240
Colpaert FC, Niemegeers CJE, Janssen PAJ (1979) Discriminative stimulus properties of cocaine: neuropharmacological characteristics as derived from stimulus generalization experiments. Pharmacol Biochem Behav 10:535–546
Cunningham KA, Appel JB (1986) Possible 5-hydroxytryptamine1 (5-HT1) receptor involvement in the stimulus properties of 1-(m-Trifluoromethylphenyl) piperizeine (TFMPP). J Pharmacol Exp Ther 237:369–377
Cunningham KA, Callahan PM (1991) Monoamine uptake inhibitors enhance the discriminative state induced by cocaine in the rat. Psychopharmacology 104:177–180
Cunningham KA, Callahan PM, Appel JB (1987) Discriminative stimulus properties of 8-hydroxy-2-(di-n-propylamino) tetralin (8-OH DPAT): implications for understanding the actions of novel anxiolytics. Eur J Pharmacol 138:29–36
Daniell HB, Pierre AJ, Webb JG (1976) A positive inotropic effect of propranolol on the canine myocardium: a presynaptic effect. J Pharmacol Exp Ther 196:657–664
Davies D, Reid J (1975) Central action of drugs in blood pressure regulation. University Park Press. Baltimore MD
Day MD, Roach AG (1974) Cardiovascular effects of beta-adrenoreceptor blocking agents after intracerebroventricular administration in conscious normotensive cats. Clin Exp Pharmacol Physiol 1:333–339
Engel G, Göthert M, Hoyer D, Schlicker E, Hillenbrand K (1986) Identity of inhibitory presynaptic 5-hydroxytryptamine (5-HT) autoreceptors in the rat brain cortex with 5-HT1B binding sites. Naunyn-Schmiedeberg’s Arch Pharmacol 332:1–7
Finney D (1952) Probit analysis. Cambridge University Press, London, UK
Fitzgerald JD (1980) Propranolol. In: Scriabine A (ed) Pharmacology of antihypertensive drugs. Raven, New York, pp 195–208
Fraundorfer PF, Fertel RH, Miller DD, Feller DR (1994) Biochemical and pharmacological characterization of high-affinity trimetoquinol analogs on guinea pig and human β-adrenergic receptor subtypes: evidence for partial agonism. J Pharmacol Exp Ther 270:665–674
Frishman WH (1988) β-Adrenergic receptor blockers: adverse effects and drug interactions. Hypertension 11:II21–II29
Frishman WH, Razin A, Swencionis C, Sonnenblick EH (1981) β-adrenoceptor blockade in anxiety states: a new approach to therapy? Cardiovasc Rev Rep 2:447–459
Gardner CR (1989) The discriminative stimulus properties of the 5-HT1 agonist RU 24969. Pharmacol Biochem Behav 33:761–764
Glennon RA (1986) Discriminative stimulus properties of the 5-HT1A agonist 8-hydroxy-2-(di-n-propylamino) tetralin (8-OH DPAT). Pharmacol Biochem Behav 25:135–139
Glennon RA, McKenney JD, Young R (1984) Discriminative stimulus properties of the serotonin agonist 1-(3-Trifluoromethylphenyl)piperazine (TFMPP). Life Sci 35:1475–1480
Glennon RA, Pierson ME, McKenney JD (1988) Stimulus generalization of 1-(3-Trifluoromethylphenyl)piperazine (TFMPP) to propranolol, pindolol, and mesulergine. Pharmacol Biochem Behav 29:197–199
Glennon RA, Lee M, Rangisetty JB, Dukat M, Roth BL, Savage JE, McBride A, Rauser L, Hufelsen S, Lee DKH (2000) 2-Substituted tryptamines: agents with selectivity for 5-HT6 serotonin receptors. J Med Chem 43:1011–1018
Goldberg SR, Gonzalez FA (1976) Effects of propranolol on behavior maintained under fixed-ratio schedules of cocaine injection or food presentation in squirrel monkeys. J Pharmacol Exp Ther 198:626–634
Gonasun LM, Langrall H (1982) Adverse reactions to pindolol administration. Am Heart J 104:482–486
Goodwin GM, Green AR (1985) A behavioral and biochemical study in mice and rats of putative selective agonists and antagonists for 5-HT1 and 5-HT2 receptors. Br J Pharmac 84:743–753
Gozlan H, El Mestikawy S, Pichat L, Glowinski J, Hamon M (1983) Identification of presynaptic serotonin autoreceptors using a new ligand. Nature 305:140–142
Hamon M, Lanfumey L, el Mestikawy S, Boni C, Miquel MC, Bolaños F, Schechter L, Gozlan H (1990) The main features of central 5-HT1 receptors. Neuropsychopharmacology 3:349–360
Harris GC, Hedaya MA, Pan W-J, Kalivas P (1996) Beta-adrenergic antagonism alters the behavioral and neurochemical responses to cocaine. Neuropsychopharm 14:195–204
Hayes A, Cooper RG (1971) Studies on the absorption, distribution and excretion of propranolol in rat, dog and monkey. J Pharmacol Exp Ther 176:302–311
Herndon JL, Pierson ME, Glennon RA (1992) Mechanistic investigation of the stimulus properties of 1-(3-Trifluoromethylphenyl)piperazine. Pharmacol Biochem Behav 43:739–748
Herrick-Davis K, Grinde E, Gauthier C, Teitler M (1998) Pharmacological characterization of the constitutively activated state of the serotonin 5-HT2C receptor. Ann N Y Acad Sci 861:140–145
Ho BT, McKenna M (1978) Discriminative stimulus properties of central stimulants. In: Ho BT, Richards DW III, DL Chute DL (eds) Drug discrimination and state dependent learning. Academic, New York, pp 67–77
Hoyer D, Engel G, Kalkman HO (1985a) Characterization of the 5-HT1B recognition site in rat brain: binding studies with [125I]-iodocyanopindolol. Eur J Pharmacol 118:1–12
Hoyer D, Engel G, Kalkman HO (1985b) Molecular pharmacology of 5-HT1 and 5-HT2 recognition sites in rat and pig brain membranes: radioligand binding studies with [3H]5-HT, [3H}8-OH DPAT, [125I]-iodocyanopindolol, [3H]mesulergine, and [3H]ketanserin. Eur J Pharmacol 118:13–23
Hoyer D, Waeber C, Schoeffter P, Palacios JM, Dravid A (1989) 5-HT1C receptor-mediated stimulation of inositol phosphate production in pig choriod plexus. A pharmacological characterization. Nauunyn Schmiedeberg’s Arch Pharmacol 339:252–258
Jarbe TUC, Ohlin GCH (1977) Stimulus effects of Δ9-THC and its interaction with naltrexone and catecholamine blockers in rats. Psychopharmacology 54:193–195
Johanson C-E, Barrett JE (1993) The discriminative stimulus effects of cocaine in pigeons. J Pharmacol Exp Ther 267:1–8
Kanba S, Richelson E (1984) Histamine H1 receptors in human brain labeled with [3H}doxepin. Brain Res 304:1–7
Kelliher GJ, Buckley JP (1970) Central hypotensive activity of dl- and d-propranolol. J Pharm Sci 59:1276–1280
Khorana N, Pullaguria MR, Young R, Glennon (2004) Comparison of the discriminative stimulus effects of 3,4-methylenedioxymethamphetamine (MDMA) and cocaine: asymmetric generalization. Drug Alcohol Depend 74:281–287
Khorana N, Young R, Glennon RA (2008) Effect of 8-Hydroxy-2-(N,N-di-n-propylamino)tetralin and MDMA on the discriminative stimulus effects of the classical hallucinogen DOM in rats. Pharmacol Biochem Behav (in press)
Kleven MS, Koek W (1997) Discriminative stimulus properties of cocaine: enhancement by β-adrenergic receptor antagonists. Psychopharmacology 131:307–312
Kleven MS, Koek W (1998) Discriminative stimulus properties of cocaine: enhancement by monoamine reuptake blockers. J Pharmacol Exp Ther 284:1015–1025
Kleven MS, Kamenka J-M, Vignon J, Koek W (1999) Pharmacological characterization of the discriminative stimulus properties of the phencyclidine analog, N-[1-(2-benzo(b)thiophenyl)-cyclohexyl]piperidine. Psychopharmacology 145:370–377
Kobinger W (1978) Central α-adrenergic systems as targets for hypotensive drugs. Rev Physiol Biochem Pharmacol 81:39–100
Koe BK (1976) Molecular geometry of inhibitors of the uptake of catecholamines and serotonin in synaptosomal preparations of rat brain. J Pharmacol Exp Ther 199:649–661
Lyon RA, Titeler M (1988) Pharmacology and biochemistry of the 5-HT2 receptor. In: Sanders-Bush E (ed) The serotonin receptors. The Humana, Clifton, pp 59–88
McKenney JD, Glennon RA (1986) TFMPP may produce its stimulus effects via a 5-HT1B mechanism. Pharmacol Biochem Behav 24:43–47
Middlemiss DN (1984) Stereoselective blockade at [3H]5-HT binding sites and at the 5-HT autoreceptor by propranolol. Eur J Pharmacol 101:289–293
Middlemiss DN, Buxton DA, Greenwood DT (1981) β-adrenoceptor antagonists in psychiatry and neurology. Pharmacol Ther 12:419–437
Munzar P, Goldberg SR (1999) Noradrenergic modulation of the discriminative-stimulus effects of methamphetamine in rats. Psychopharmacology 143:293–301
Neil-Dwyer G, Bartlett J, McAinsh J, Cruickshank JM (1981) β-adrenoceptor blockers and the blood–brain barrier. Br J Clin Pharmacol 11:549–553
O’Donnell JM (1997) Pharmacological characterization of the discriminative stimulus effects of clenbuterol in rats. Pharmacol Biochem Behav 58:813–818
Offord SJ, Ordway GA, Frazer A (1988) Application of [125]iodocyanopindolol to measure 5-hydroxytryptamine1B receptors in the brain of the rat. J Pharmacol Exp Ther 244:144–153
Ordway WA, O’Donnell JM, Frazer A (1987) Effects of clenbuterol on central beta1- and beta2-adrenergic receptors in the rat. J Pharmacol Exp Ther 241:187–195
Peroutka SJ (1986) Pharmacological differentiation and characterization of 5-HT1A, 5-HT1B, 5-HT1C binding sites in rat frontal cortex. J Neurochem 47:529–540
Petrie WM, Maffucci RJ, Woosley RL (1982) Propranolol and depression. Am J Psychiatry 139:92–94
Pierson ME, Lyon RA, Titeler M, Kowalski P, Glennon RA (1989) Design and synthesis of propranolol analogues as serotonergic agents. J Med Chem 32:859–863
Privitera PJ, Webb JG, Walle T (1979) Effects of centrally administered propranolol on plasma renin activity, plasma norepinephrine and arterial pressure. Eur J Pharmacol 54:51–60
Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS (2001) Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 39:32–41
Rowlett JK, Platt DM, Spealman RD (2004) Cocaine-like discriminative stimulus effects of heroin: modulation by selective monoamine transport inhibitors. J Pharmacol Exp Ther 310:342–348
Sadzot B, Baraban JM, Glennon RA, Lyon RA, Leonhardt S, Jan CR, Titler M (1989) Hallucinogenic drug interactions at human brain 5-HT2 receptors: implications for treating LSD-induced hallucinogenesis. Psychopharmacology 98:495–499
Saelens DA, Daniell HB, Webb JG (1977) Studies on the interactions of propranolol with adrenergic neurons. J Pharmacol Exp Ther 202:635–645
Schechter MD (1974) Effect of propranolol, d-amphetamine and caffeine on ethanol as a discriminative cue. Eur J Pharmacol 29:52–57
Schechter MD, Cook PG (1975) Dopaminergic mediation of the interoceptive cue produced by d-amphetamine in rats. Psychopharmacologia 42:185–193
Schlegel JR, Peroutka SJ (1986) nucleotide interactions with 5-HT1A binding sites directly labeled by [3H]-8-hydroxy-2-(Di-n-propylamino)tetralin ([3H]-8-OH-DPAT). Biochem Pharmacol 35:1943–1949
Schlicker E, Fink K, Göthert M, Hoyer D, Molderings G, Roschke I, Schoeffter P (1989) The pharmacological properties of the presynaptic serotonin autoreceptor in the pig brain cortex conform to the 5-HT1D receptor subtype. Naunyn Schmiedebergs Arch Pharmacol 340:45–51
Schotte A, Janssen PF, Gommeren W, Luyten WH, Van Gompel P, Lesage AS, De Loore K, Leysen JE (1996) Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology 124:57–73
Simpson FO (1974) β-adrenergic receptor blocking drugs in hypertension. Drugs 7:85–105
Spealman RD (1995) Noradrenergic involvement in the discriminative stimulus effects of cocaine in squirrel monkeys. J Pharmacol Exp Ther 275:53–62
Spencer DG, Glaser T, Traber J (1987) Serotonin receptor subtype mediation of the interoceptive discriminative stimuli induced by 5-methoxy-N,N-dimethyltryptamine. Psychopharmacology 93:158–166
Sprouse JS, Aghajanian GK (1986) (−)-Propranolol blocks the inhibition of serotonergic dorsal raphe cell firing by 5-HT1A selective agonists. Eur J Pharmacol 128:295–298
Tackett RL, Webb JG, Privitera PJ (1981) Cerebroventricular propranolol elevates cerebrospinal fluid norepinephrine and lowers blood pressure. Science 213:911–913
Terry P, Witkin JM, Katz JL (1994) Pharmacological characterization of the novel discriminative stimulus effects of a low dose of cocaine. J Pharmacol Exptl Ther 270:1041–1048
To ZP, Bonhaus DW, Eglen RM, Jakeman LB (1995) Characterization and distribution of putative 5-HT7 receptors in guinea-pig brain. Br J Pharmacol 115:107–116
Toll L, Berzetei-Gurske IP, Polgar WE, Brandt SR, Adapa ID, Rodriguez L, Schwartz RW, Haggart D, O’Brien A, White A, Kennedy JM, Craymer K, Farrington L, Auh JS (1998) Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Res Monogr 178:440–466
Tricklebank MD (1985) The behavioural response to 5-HT receptor agonists and subtypes of the central 5-HT receptor. Trends Pharmacol Sci 5:403–407
Tricklebank MD, Forler C, Fozard JR (1985) The involvement of subtypes of the 5-HT1 receptor and of catecholaminergic systems in the behavioural response to 8-hydroxy-2-(di-n-propylamino)tetralin in the rat. Eur J Pharmacol 106:271–282
Tricklebank MD, Neill J, Kidd EJ, Fozard JR (1987) Mediation of the discriminative stimulus properties of 8-hydroxy-2-(di-n-propylamino)tetralin (8-OH DPAT) by the putative 5-HT1A receptor. Eur J Pharmacol 133:47–56
Tsuchihashi H, Nakashima Y, Kinami J, Nagatomo T (1990) Characteristics of 125I-iodocyanopindolol binding to beta-adrenergic and serotonin-1B receptors of rat brain: selectivity of beta-adrenergic agents. Jpn J Pharmacol 52:195–200
Tuross N, Patrick RL (1986) Effects of propranolol on catecholamine synthesis and uptake in the central nervous system of the rat. J Pharmacol Exp Ther 237:739–745
Wainscott DB, Lucaites VL, Kursar JD, Baez M, Nelson DL (1996) Pharmacologic characterization of the human 5-hydroxytryptamine2B receptor: evidence for species differences. J Pharmacol Exp Ther 276:720–727
Winter JC (1975) The stimulus properties of morphine and ethanol. Psychopharmacologia 44:209–214
Woods PB, Robinson ML (1981) An investigation of the comparative liposolubilities of β-adrenoceptor blocking agents. J Pharm Pharmacol 33:172–173
Young R, Glennon RA (1986) Amphetamine and related phenylalkylamines as discriminative stimuli: a review of the literature. Med Res Rev 6:99–130
Young R, Glennon RA (1988) Second generation anxiolytics and serotonin. In: Rech RH, Gudelsky GA (eds) 5-HT agonists as psychoactive drugs. NPP Books, Ann Arbor, pp 239–258
Young R, Glennon RA (1997) Cocaine-stimulus generalization to MDA optical isomers: a re-evaluation. Pharmacol Biochem Behav 57:115–118
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was supported, in part, by the National Institute on Drug Abuse (NIDA) grant DA-01642.
Rights and permissions
About this article
Cite this article
Young, R., Glennon, R.A. S(−)Propranolol as a discriminative stimulus and its comparison to the stimulus effects of cocaine in rats. Psychopharmacology 203, 369–382 (2009). https://doi.org/10.1007/s00213-008-1317-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00213-008-1317-2