Reactome can annotate and display pathways associated with disease. Reactome disease annotations include cancer, metabolic and immune disease as well as infectious diseases, among others. Where possible, Reactome disease pathways also include the interaction of relevant therapeutic drugs. (Disease pathways are contained in a separate top-level Chapter in the hierarchy, and are designated with a red “+” symbol to the left of the pathway name.)
Pathway Diagrams:
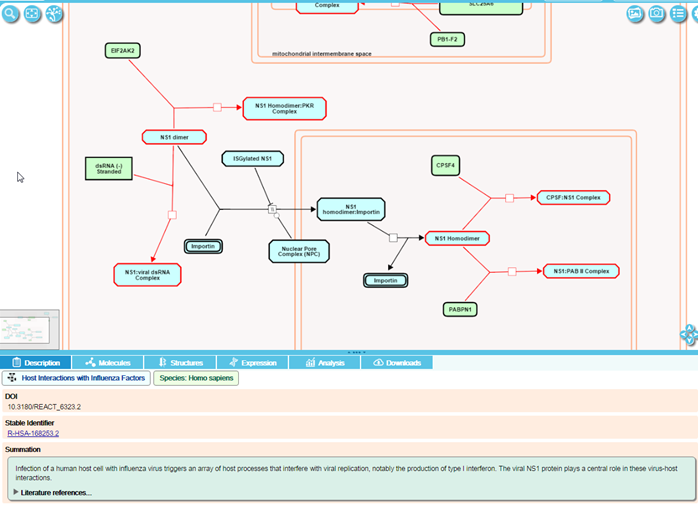
Disease events or processes are shown in the context of the normal pathway diagram to provide context to the disease event.
Disease events have red connecting lines; abnormal molecules involved in these processes are outlined in red.
For infection processes, events involved in the life cycle of the infecting agent and host interactions with the infecting agent have red connecting lines. Molecules derived from the infecting agent are outlined in red, while those derived from the host are outlined as normal in black.
Disease events may be loss-of-function or gain-of-function compared to the normal process.
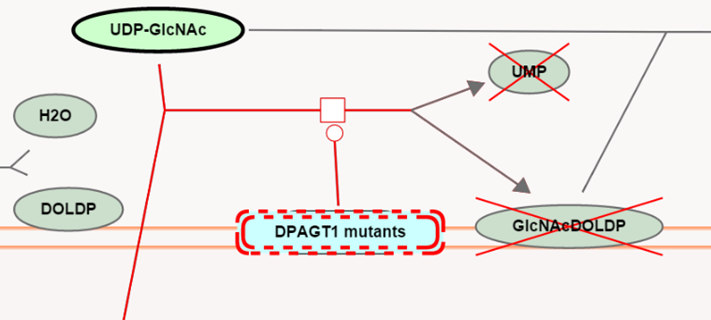
For loss-of-function events (where a protein has lost all or most of its functional activity) the disease reaction is automatically overlaid on top of the corresponding normal reaction. The loss-of-function disease entity is outlined with a dashed red line, and products that are no longer made are greyed out with a superimposed red cross. These loss-of-function events represent “stop points” in the pathway.
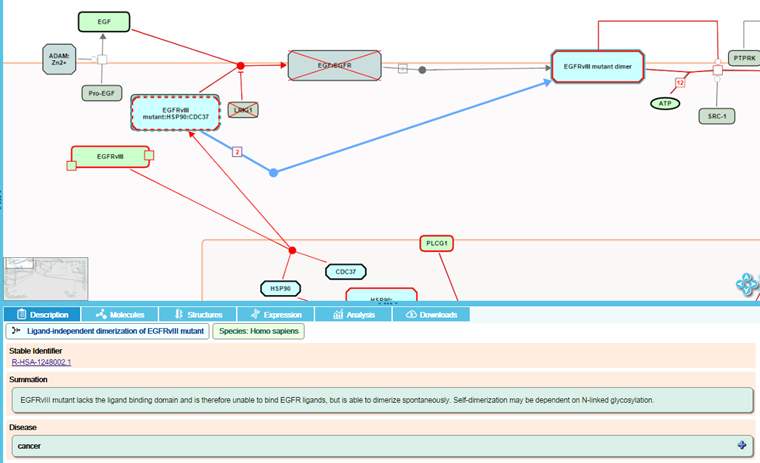
For gain-of-function events (where a protein has acquired a novel function not performed by the wild-type protein), disease events are represented alongside the normal events.
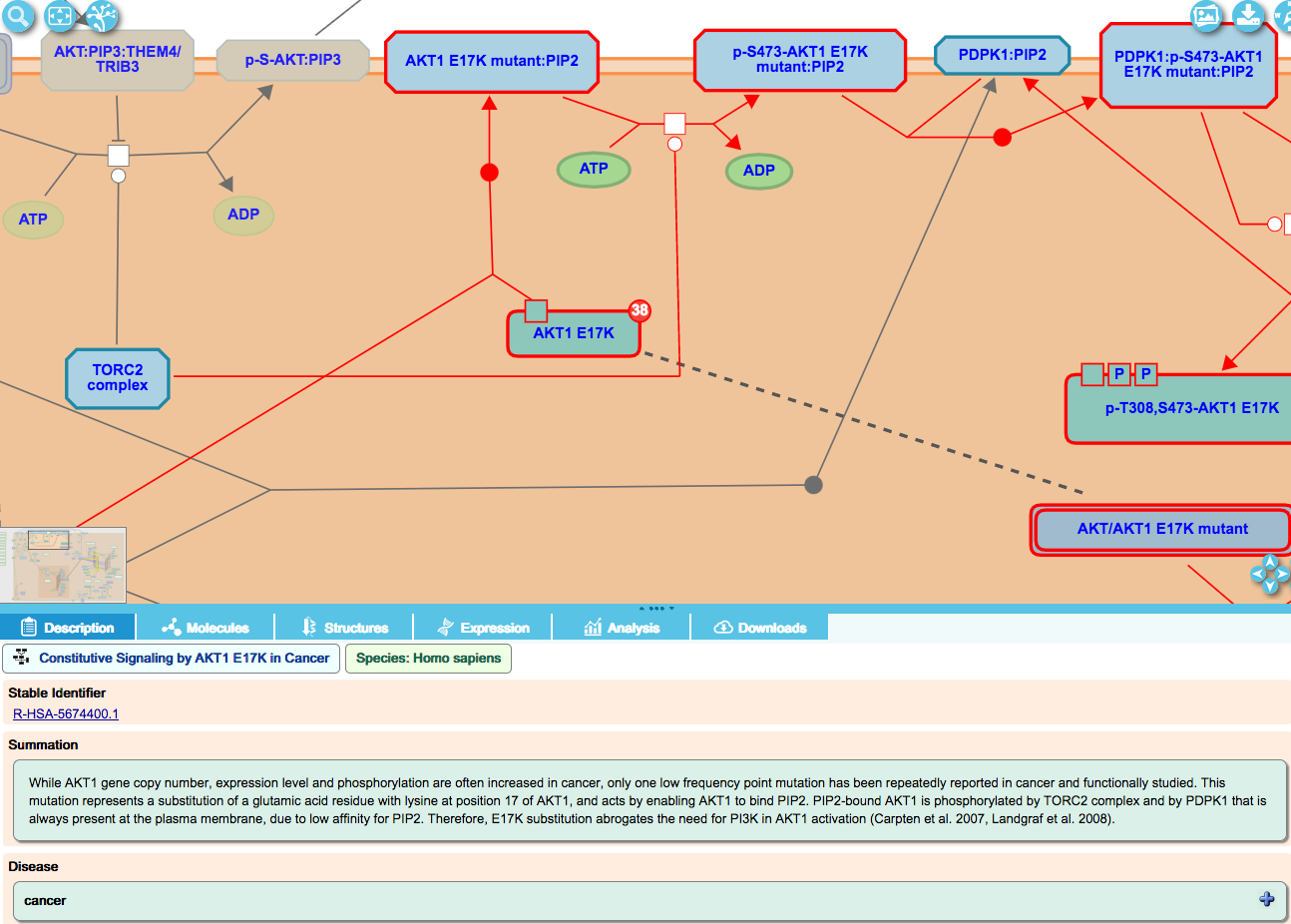
When a gain-of-function mutant performs a normal function (either a higher rate or efficiency, or after being activated in an novel manner, for instance), these disease events are overlaid on the corresponding normal event in the pathway diagram.
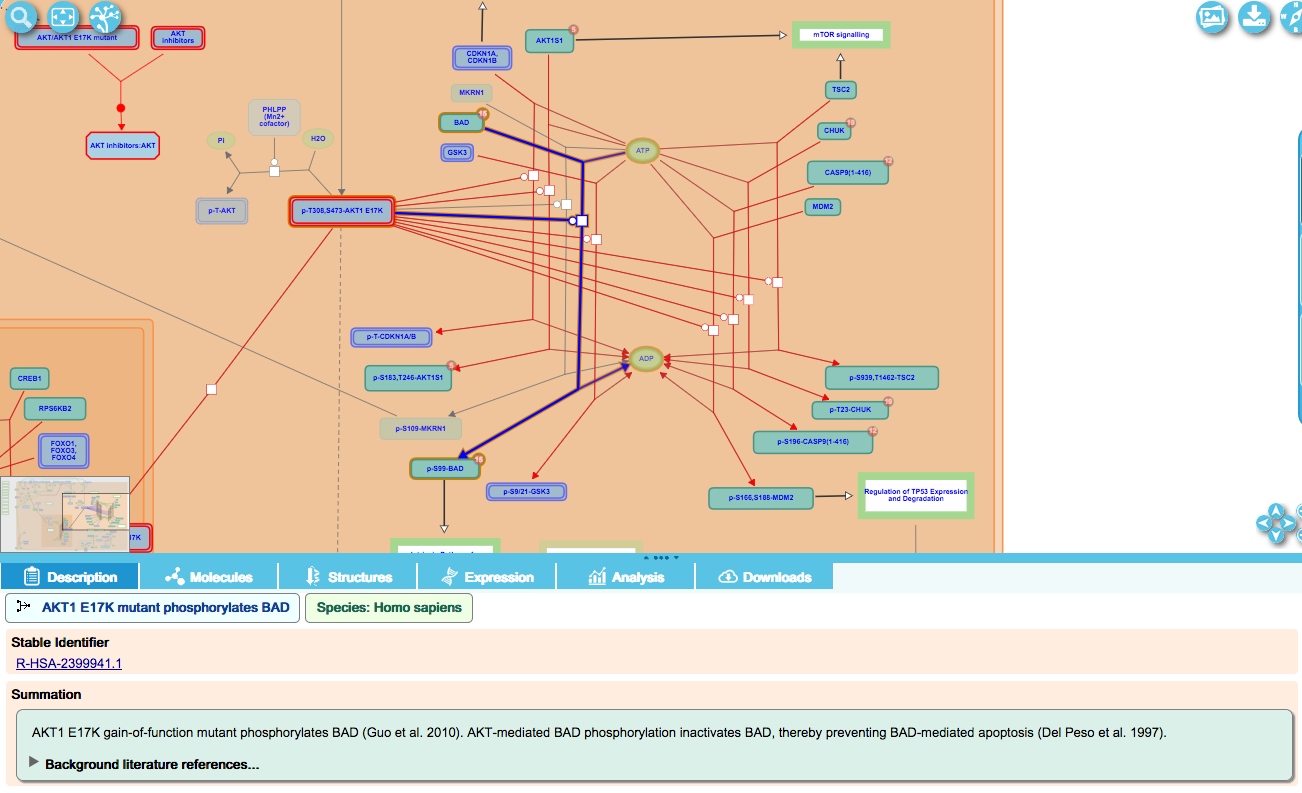
Infectious disease events (which don’t occur in the absence of the infectious agent and therefore have no normal counterpart) have their own diagrams.
Drug annotations:
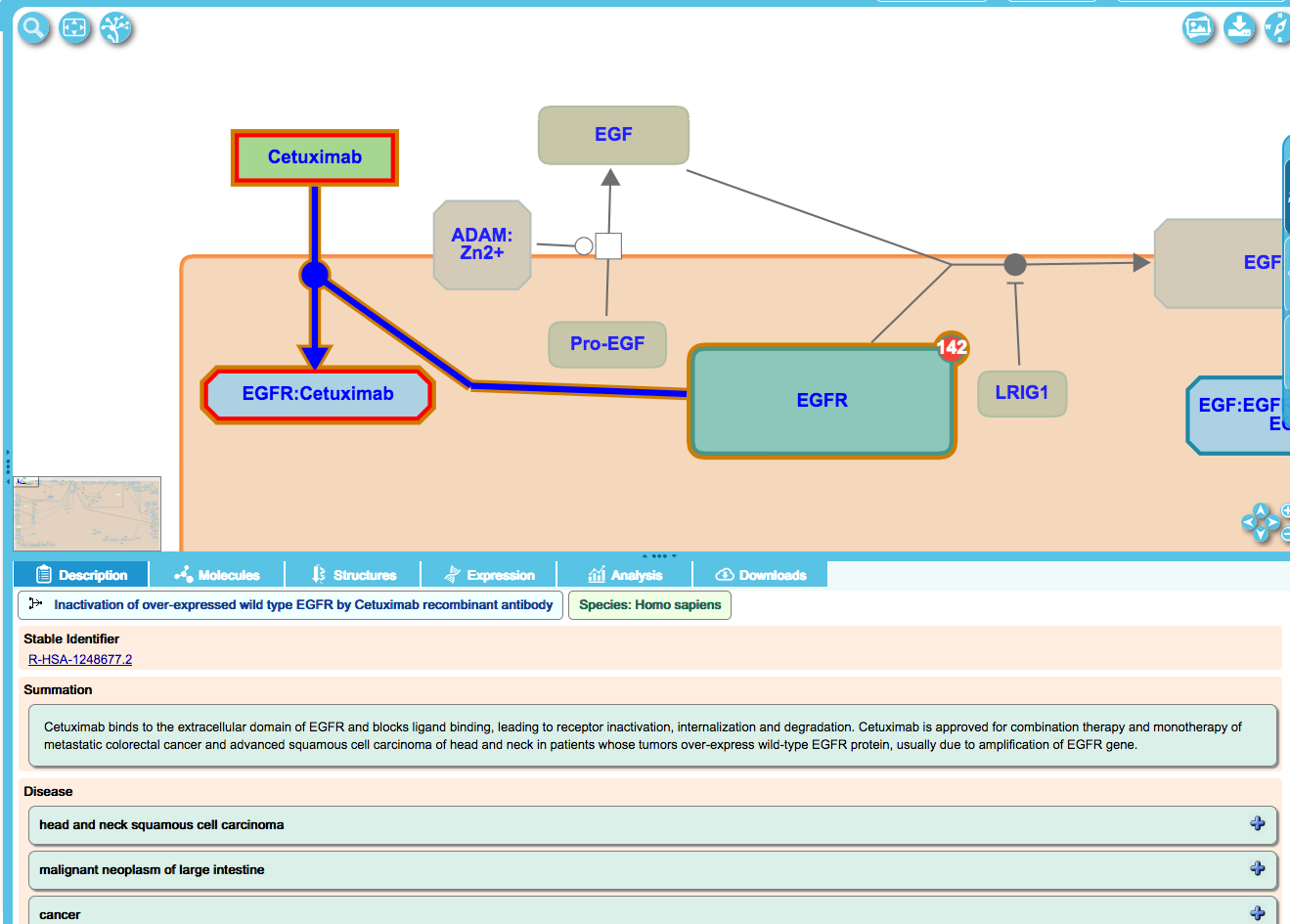
Where applicable, the effect of drugs on disease pathways is annotated.
Details panel:
The details panel for a disease pathway/event has a number of extra panels in addition to those displayed in a normal pathway/event.
Disease tag:
Disease events and entities are tagged with a disease term, taken from the Disease Ontology and displayed in the “Disease” panel.
Individual proteins are labeled with all the relevant disease terms, while sets of disease entities are tagged with the most specific term that represents all of the set members.

Where possible, disease proteins are linked to OMIM (Online Inheritance in Man),
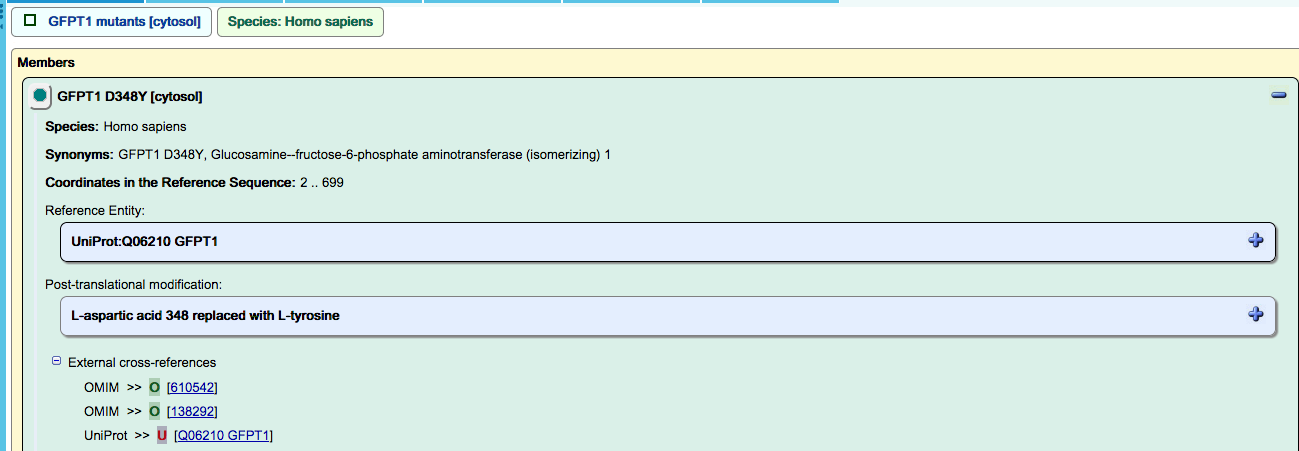
and cancer variants are linked to COSMIC (Catalogue of Somatic Mutations in Cancer).

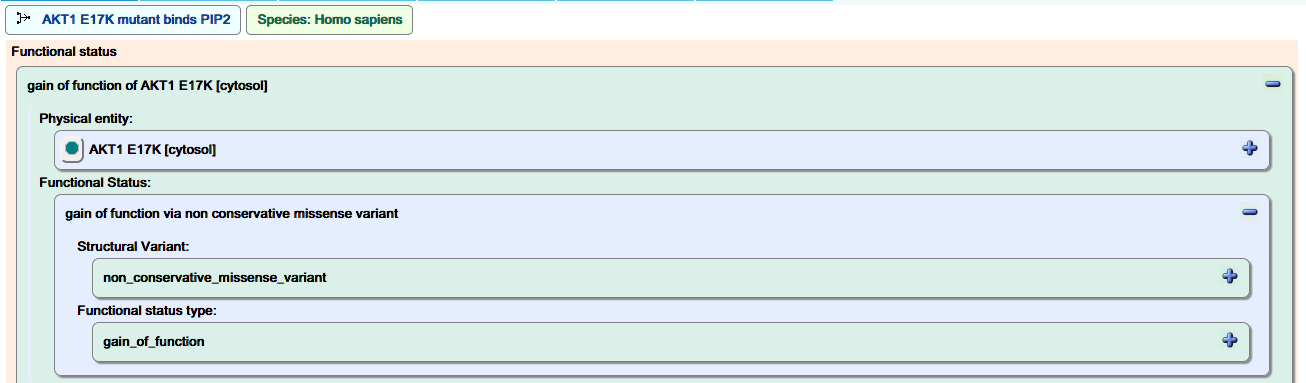
Functional status:
As described above, disease events are tagged as either gain- or loss-of-function, and this is displayed in the “Functional Status” panel.
Normal Pathway/Reaction:
Where applicable, the corresponding normal pathway or reaction is identified in the details panel of the disease event.

Disease entities:
Disease entities are annotated in molecular detail. Changes to protein sequence that arise as a result of variation in DNA sequence in disease are displayed in the details panel when a disease entity is highlighted in the diagram. Missense, nonsense, frameshift, deletion and fusion mutations are all annotated. See “More info”, below, for details of these annotations.
Primary references describing the identification of the mutants proteins are annotated, but this information is not currently displayed on the website. Note also that on the website, these genetic modifications are displayed in the same panel as post-translational modifications such as phosphorylations, and are therefore (mis)labeled as ‘Post-translational modifications’.
Missense mutation:
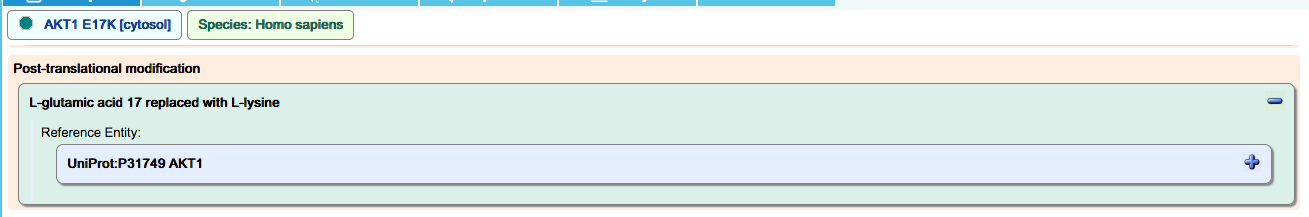
Deletion mutation:
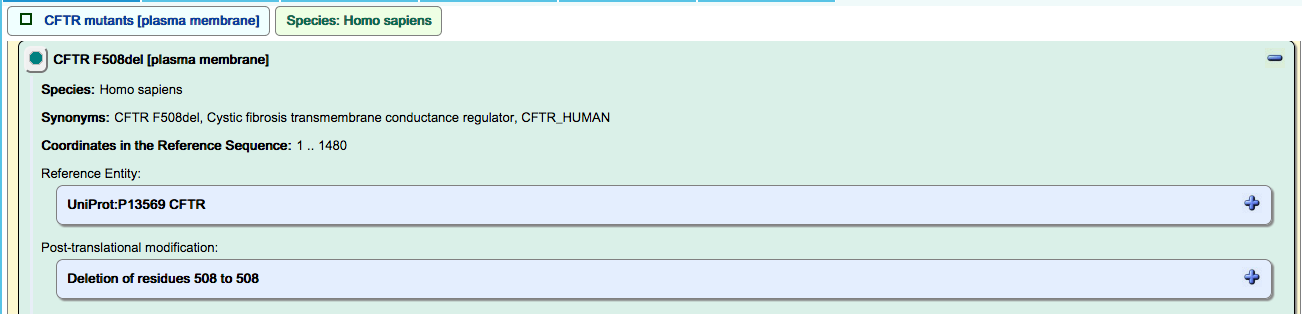
Drugs:
Drugs are cross-referenced to ChEBI and to IUPHAR where possible and applicable, and this information is displayed in the details panel when a drug is highlighted in the diagram.
Get started:
Example 1 - drug-target interaction in disease:
The cystic fibrosis transmembrane conductance regulator (CFTR) is a low conductance chloride-selective channel that mediates the transport of chloride ions in human airway epithelial cells. Chloride ions plays a key role in maintaining homeostasis of epithelial secretions in the lungs. Defects in CFTR can cause cystic fibrosis (CF), resulting in an ionic imbalance that impairs clearance of secretions, not only in the lung, but also in the pancreas, gastrointestinal tract and liver. More than 1500 mutations in the CFTR gene have been identified.
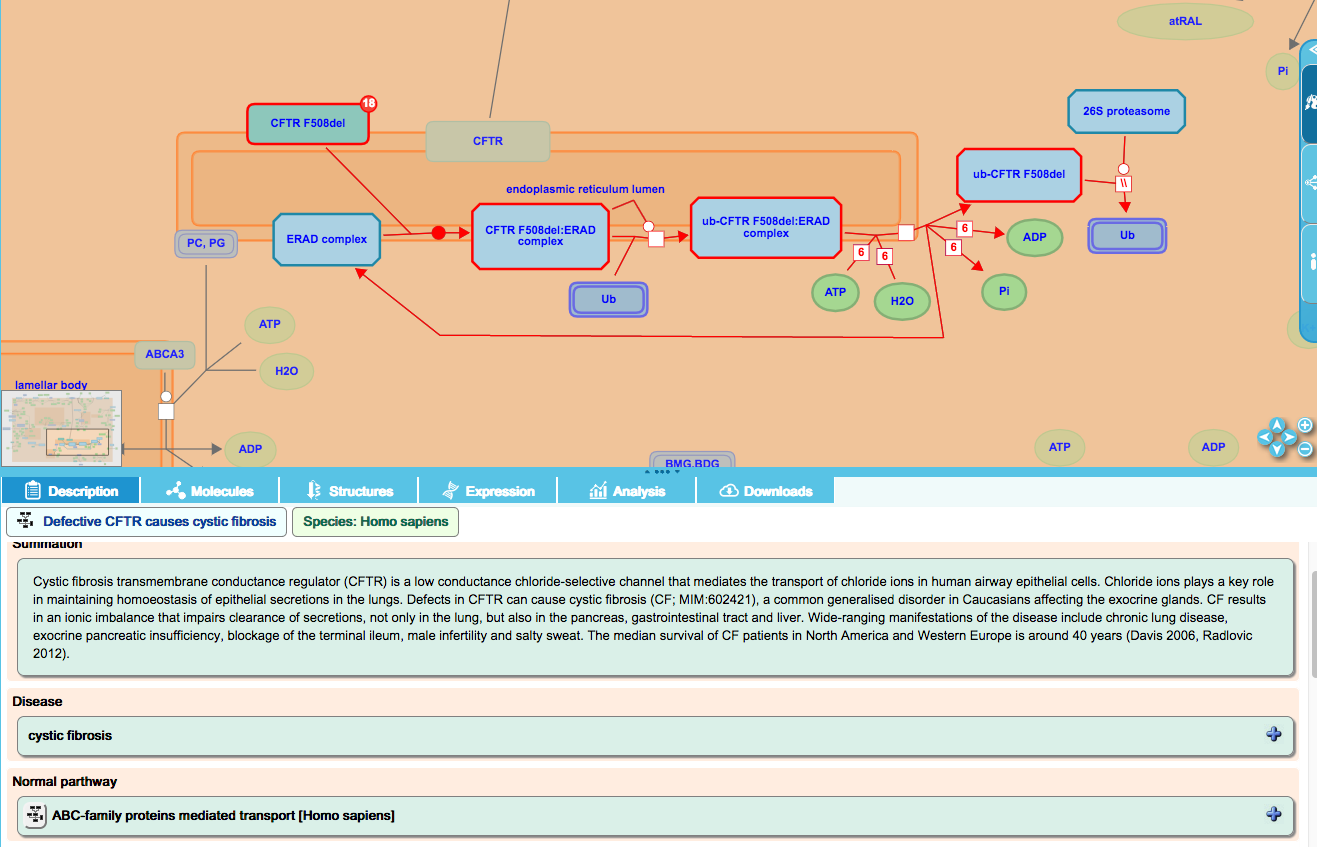
Gain-of-function events involving CFTR F508del:
Deletion of phenylalanine 508 in CFTR is the most prevalent mutation causing cystic fibrosis. F508 deletion causes destabilization and subsequent targeting for co-translational degradation by the ER-associated degradation machinery (ERAD). F508del is ubiquitinated by ERAD-associated E3 ligases including RNF5 and RNF185, targeting it for VCP-mediated retrotranslocation and 26S proteasomal degradation. These events are novel for the mutant protein and are represented in the disease diagram “ABC transporter disorders”.
Loss-of-function events for CFTR mutants:
Some loss-of-function CFTR mutants are properly transported to the plasma membrane but are unable to transport chloride ions to the extracellular space. These are represented as a loss-of-function event in an overlay of the corresponding WT reaction, and displayed in the “ABC transporter disorders” disease diagram.
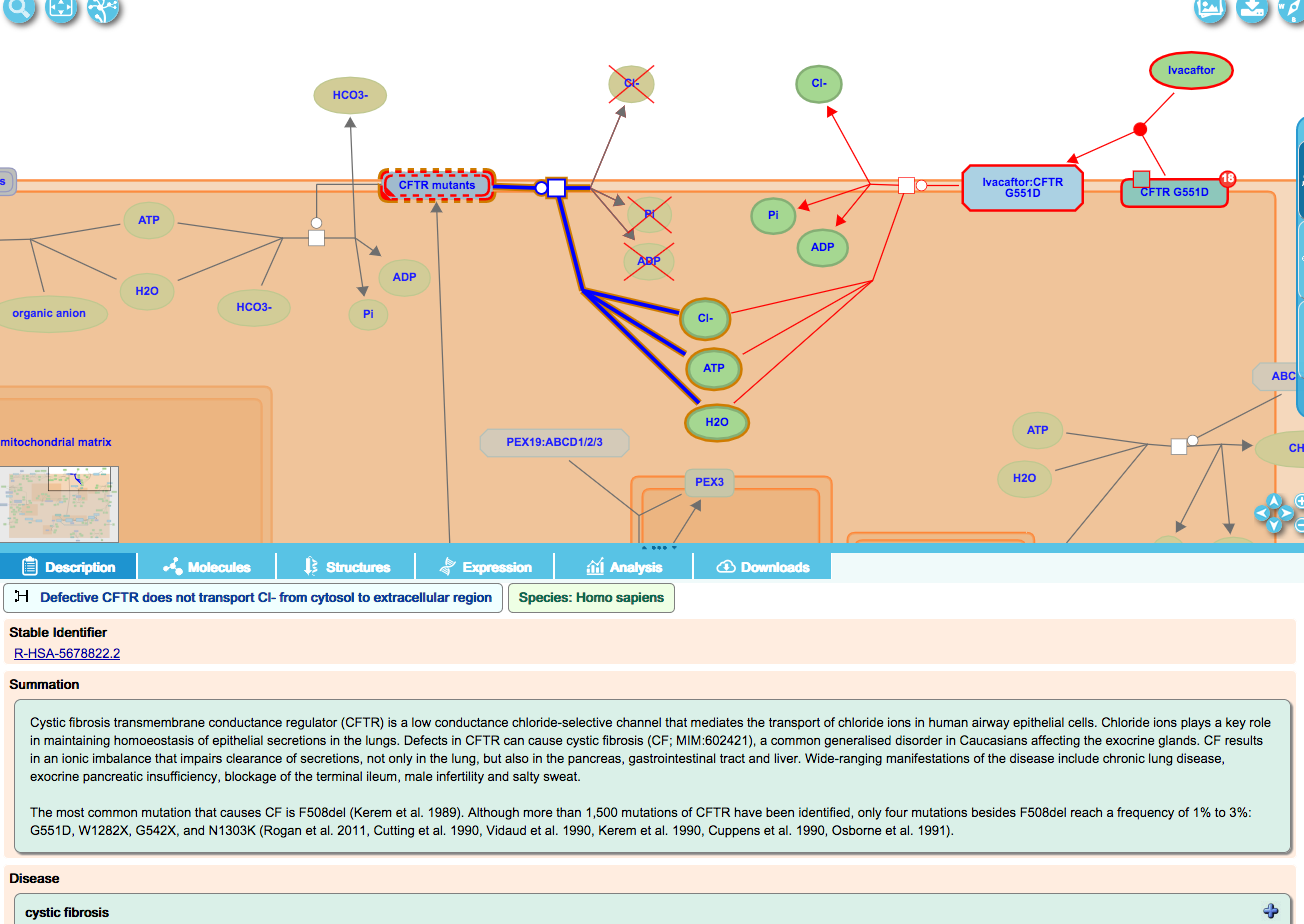
Drug interactions in Cystic Fibrosis:
CF patients with a particular mutation, G551D, have shown lung function improvements when given the drug Ivacaftor. Reactions showing ivacaftor binding the mutant protein (top right, above) and the following reaction (centre) showing channel functionality restored are displayed in the disease diagram.
Example 2 - diseases caused by accumulation of substrate over time:
Special case in Reactome where the reaction describing a particular substrate's metabolism occurs in normal physiology but over time, accumulation of the substrate leads to toxic consequences which can lead to disease. Examples are neurodegenerative diseases such as Alzheimer's and Parkinson's diseases, chronic obstructive pulmonary disease and retinal macular degeneration.
The bisretinoid A2E (di-retinoid-pyridinium-ethanolamine) is a major component of lipofuschin, a yellow-brown pigment grain composed mainly of lipids but also sugars and certain metals whose accumulation is associated with degenerative diseases. In the eye, A2E is the end-product of the condensation of 2 molecules of all-trans-retinal and phosphatidylethanolamine in photoreceptor outer disc membranes. Once formed, A2E is phagocytosed, together with outer segments, to retinal pigment epithelial (RPE) cells where it accumulates. There is no evidence as yet to indicate that A2E can be catabolised.
The relationship between lipofuscin accumulation and retinal degeneration is illustrated by Stargardt disease type 1. Because the reactions can be considered "normal" (they occur as part of the normal metabolism of retinoids) as well as disease-causing, they appear in the normal pathway diagram (Visual phototransduction) and coloured red.
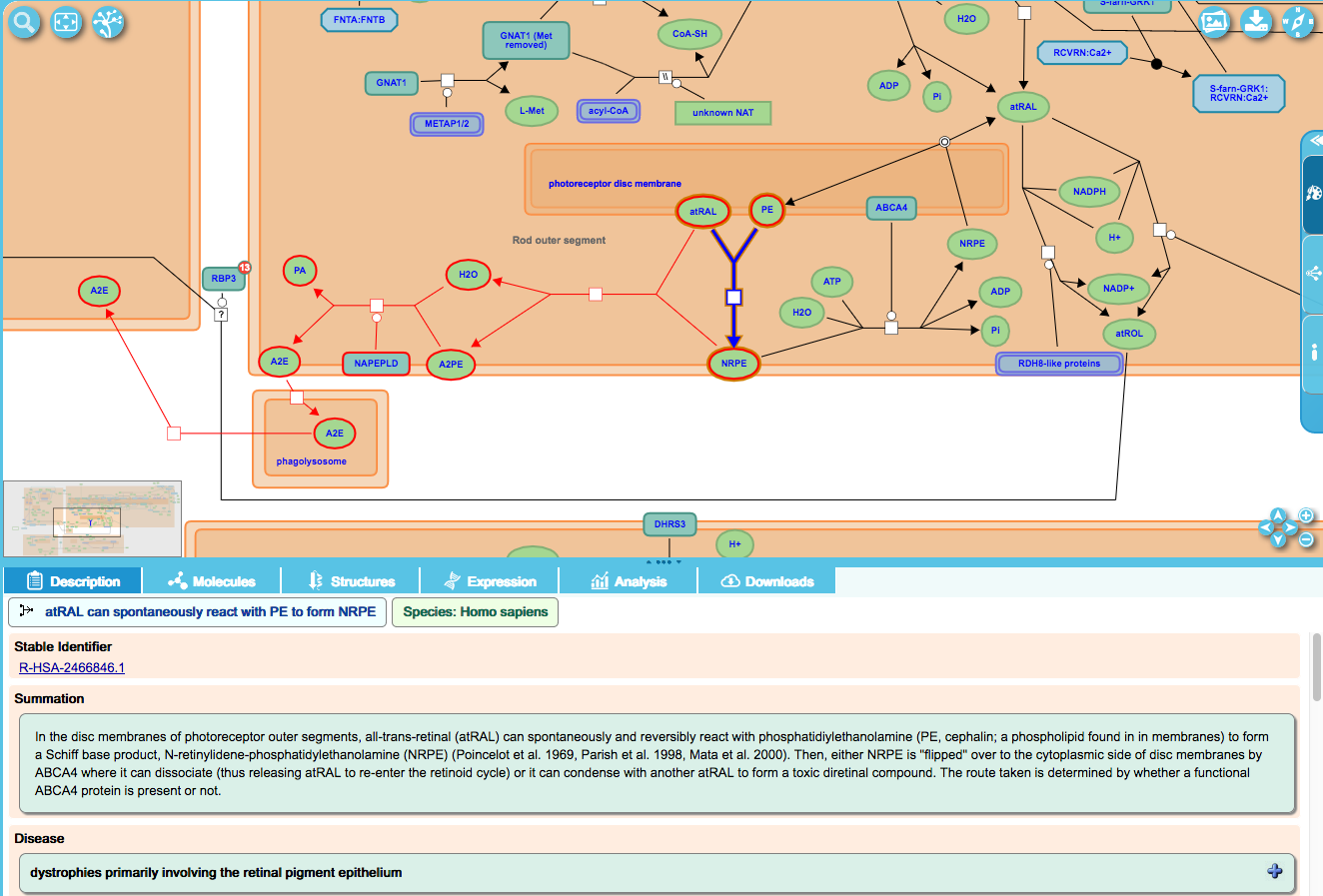
To display in the disease view of the diagram, these reactions are added as components of the disease pathway "Diseases associated with visual transduction". These reactions will now display in both normal and disease diagrams.

Getting Started
Exercises:
- Search for the pathway FGFR1 mutant receptor activation and open it. What disease(s) is/are associated with this pathway? (Hint: look at the Description tab in the Details section).
- Search for the reaction Defective MMADHC does not bind MMACHC:B12r. What type of defect has caused this loss of function? (Hint: look at the Functional Status category for the reaction).
- How many mutations of MMADHC are represented?
More info:
Annotating genetic alteration of protein sequence for disease pathways:
The following image shows the portion of the Reactome data model describing the relationship between possible modifications to protein sequence. The subclass “GeneticallyModifiedResidue” is used for the annotation of disease entities, while “TranslationalModification” is used to annotate the consequence of processes such as phosphorylation, acylation, cross-linking and other similar non-genetic events. TranslationalModifications will not be described further here.

The ReplacedResidue class is used for amino-acid substitutions. This class is also used for “simple” nonsense mutations that change a coding amino acid for a stop codon.
The FragmentModification class describes more extensive changes to the coding sequence through insertions and deletions in the gene, and includes three subclasses:
- FragmentDeletionModification is used for in-frame deletions of amino-acids
- FragmentInsertionModification is used for in-frame insertions of amino-acids, including genomic events that result in fusion proteins
- FragmentReplacedModification is used for frameshifts.
Examples of each annotation type are further described below.
Simple missense mutation: HHAT G287V
Hedgehog (Hh) is a secreted morphogen that regulates a number of developmental processes in vertebrates, including limb development and neural tube patterning, among others. Maturation of Hh ligand includes a number of proteolytic processing and lipid modification steps. These modifications are required for normal transit of the ligand to the surface of the secreting cell and mutations that affect these processes are associated with decreased Hh ligand secretion, abrogated Hh signaling and disease.
HHAT is an O-acyltransferase that palmitoylates the N-terminal fragment of Hh. A G287V loss-of-function mutation in HHAT was identified in a rare case of Syndromic 46 XY Disorder of Sex Development, which results in testis dysgenesis. This mutant is not able to palmitoylate the Hh ligand. Details of the amino-acid substitution are displayed in the details panel when the entity is highlighted in the diagram:
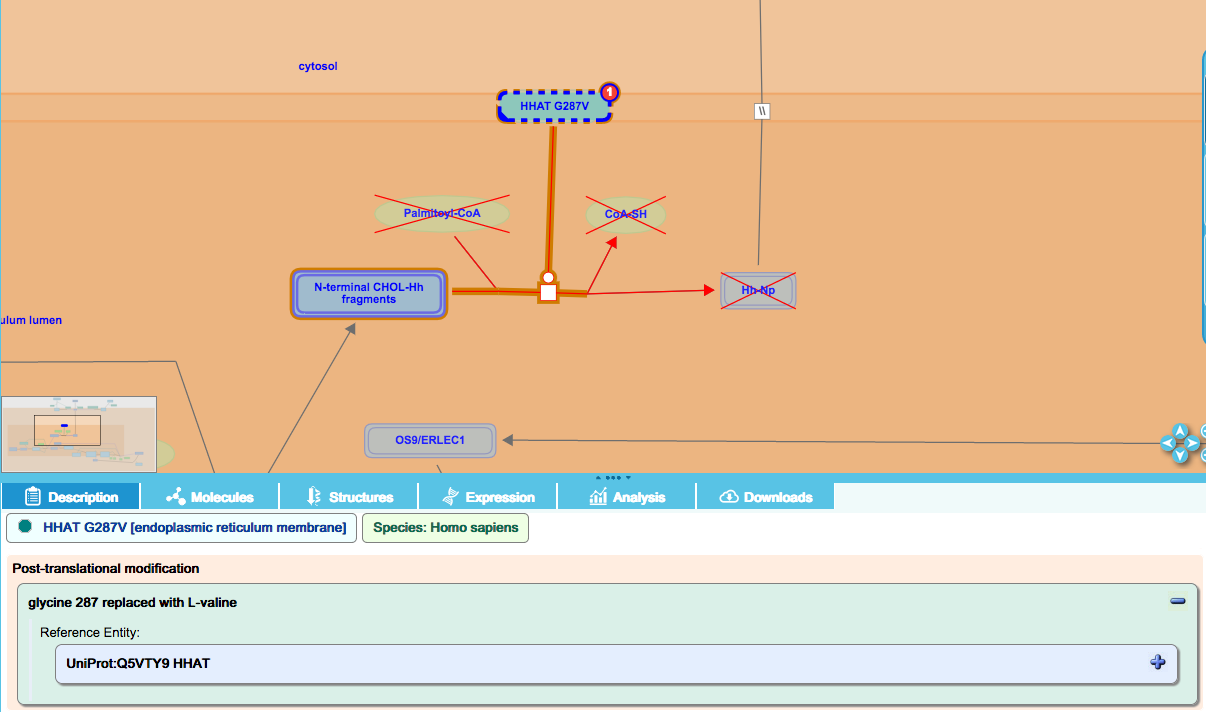
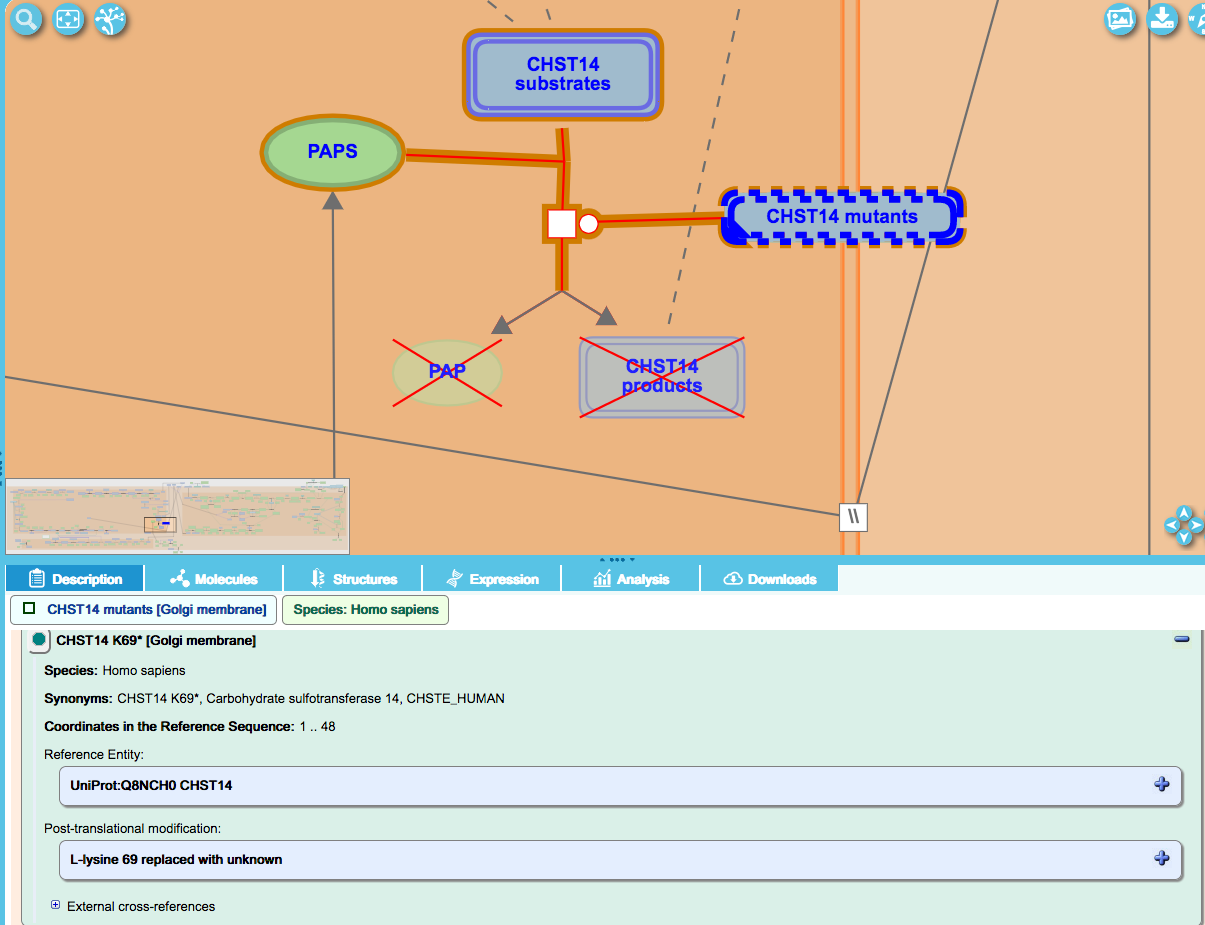
Simple nonsense mutation:
The disorder “Ehlers-Danlos syndrome, musculocontractural type 1” (EDSMC1) is caused by loss-of-function mutations in the carbohydrate sulfotransferase 14 (CHST14) gene. A nonsense mutation causing this disorder is a 205A-T transversion in the CHST14 gene, resulting in a lys69-to-ter (K69*) substitution. This is represented in the details panel as “L-lysine 69 replaced with unknown”.
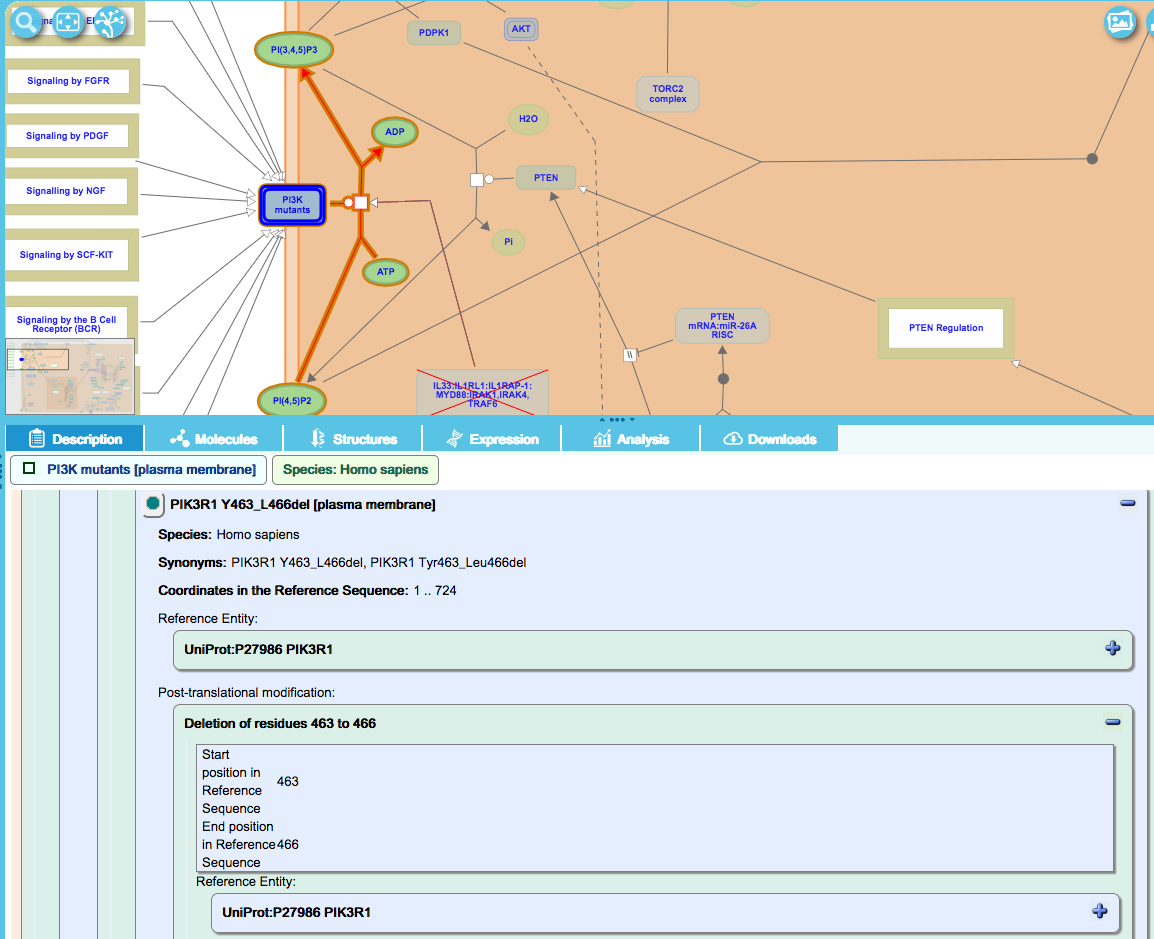
FragmentDeletionModification:
This class is used for in-frame deletions of amino-acids leading to internally truncated proteins. These variants are named “core protein name” [first amino acid of deletion_last amino acid of deletion]del, as shown below for PIK3R1 Y463_L466del, which has a deletion of residues Y463 to L466, inclusive:
FragmentInsertionModification
This class is used for in-frame insertions of amino-acids and for fusion proteins.
In-frame insertions:
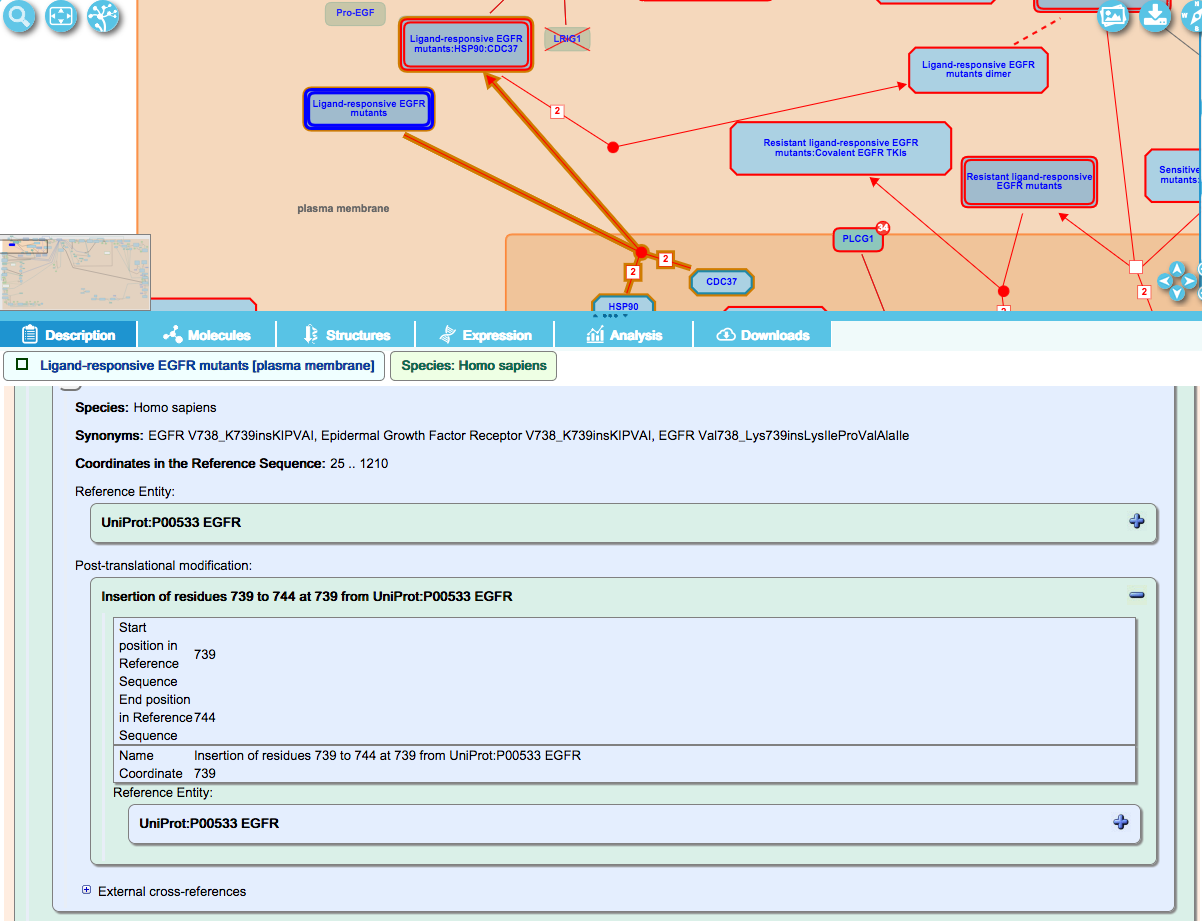
These variants are named “core protein name” [aa prior to insertion_aa following insertion]ins[inserted aa’s]. The amino-acid string of the inserted residues is added manually to the mutant protein name, as shown below for EGFR V738_K739insKIPVAI. This represents a variant where the amino acids 739-744 (KIPVAI) from EGFR are inserted at aa 739 of EGFR, and is thus a duplication of these residues:
Fusion proteins:
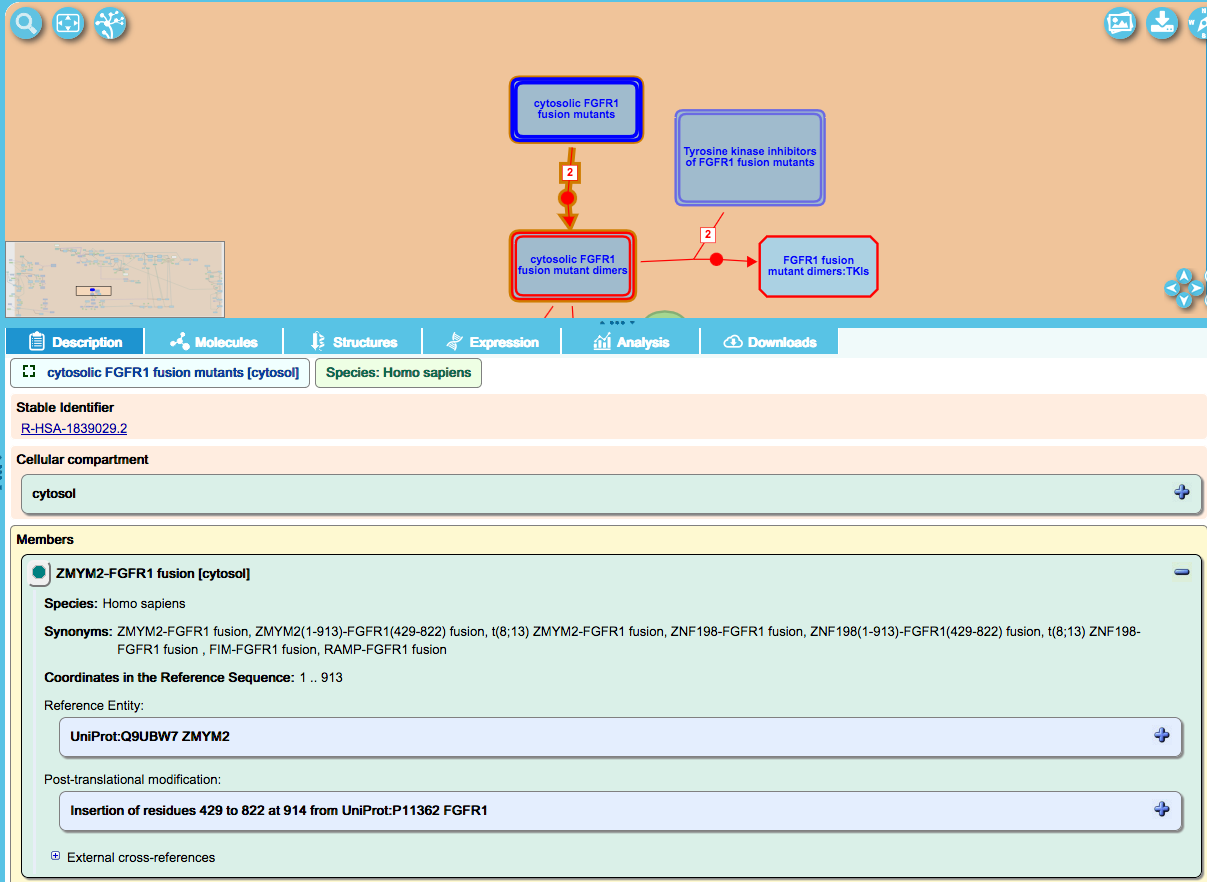
The FragmentInsertionModification class is also used to annotate proteins that arise as the result of genomic changes that bring two genes together to result in a fusion protein, as in the ZMYM2-FGFR1 fusion described below. This fusion puts the ZMYM2 dimerization region (1-914) together with the kinase domain of the FGFR1 receptor (residues 429-822) and results in constitutive activation of the kinase domain by virtue of ligand-independent dimerization (for reference, the full length aa sequence of these two proteins are 1-1377 and 1-822 for ZMYM2 and FGFR1, respectively).
By convention, the N-terminal most partner of the fusion is set as the reference protein for the variant protein while the C-terminal fusion partner sequence is captured in the FragmentInsertionModification record, as in the example below for the ZMYM2-FGFR1 fusion:
Post-translational modifications to either partner in the fusion protein are numbered according to each respective WT reference gene product and do not reflect the aa position in the fusion. For instance, if in the context of the fusion protein, the FGFR1 partner is phosphorylated at (WT FGFR1 position) Y766, the fusion EWAS would be ZMYM2-pY766-FGFR1, despite the fact that in linear sequence the phosphorylation occurs at residue 1250 of the fusion (913+(766-429)).
FragmentReplacedModification
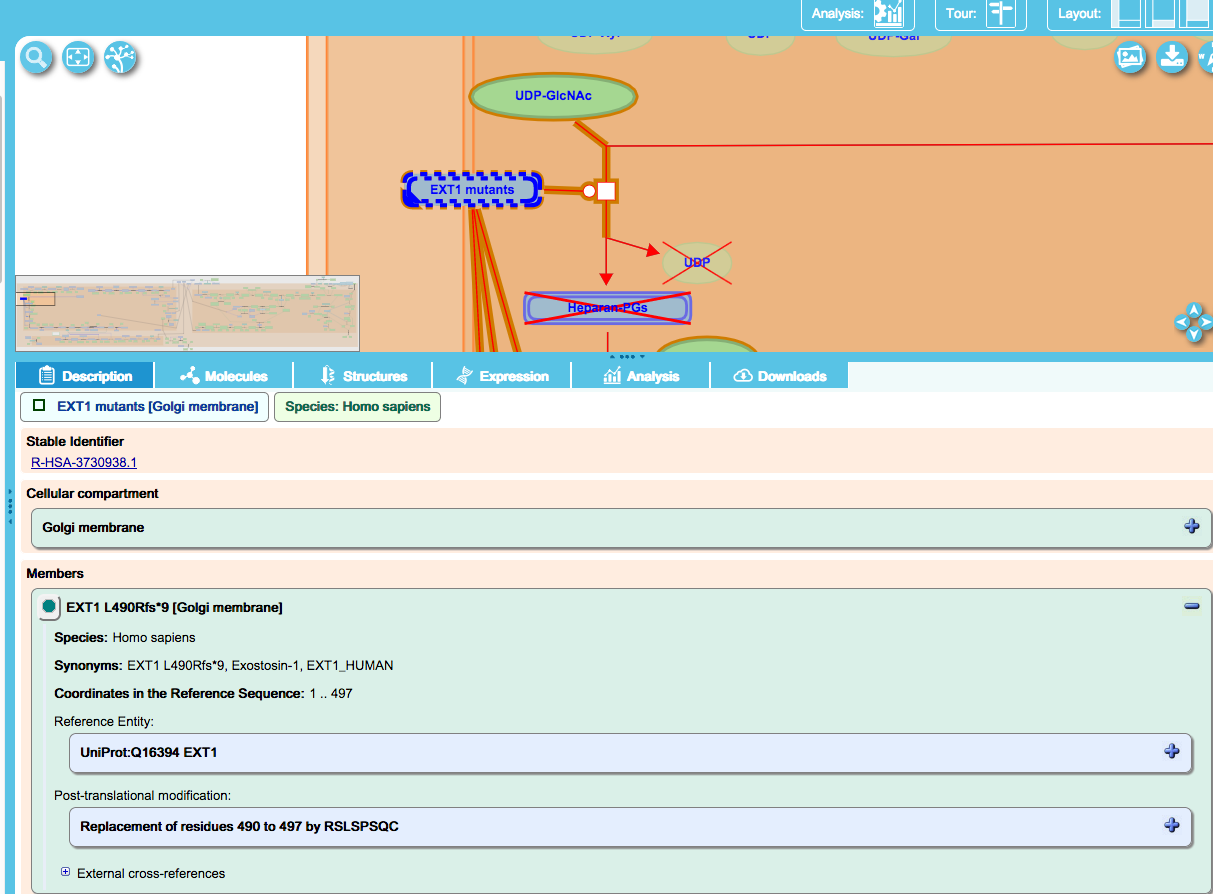
This class is used to annotate frameshift mutations that alter the amino-acid sequence of the protein as in the case of EXT1, described below.
The disorder “Hereditary multiple exostoses 1” (EXT1) is caused by loss-of-function mutations in Exostosin 1 (EXT1). One such mutation is a 1-bp deletion at nucleotide 1469 in the EXT1 gene, resulting in a frameshift mutation with a premature stop codon nine amino acids downstream. Variants of this type are named “core protein name” [aa prior to insertion_fs*(number of aa to the first stop codon)]. The novel amino acids that occur as a result of the frameshift are identified in the mutant protein record, as shown below for EXT1 L490Rfs*9:
For cancer-related processes, it is useful to view the altered disease events alongside their normal counterparts in the same diagram. The user can then see where normal processes diverge into ones that are implicated in cancer.
For metabolic processes, proteins that have lost all or most of their functional activity towards a substrate causes the majority of defects. The diagram displays these disease reactions as 'stop points' in the pathway. Defective enzyme catalysts are outlined by a red dashed line; products that are no longer made are shown greyed-out with a superimposed red cross.