

Abstract
The gray wolf (Canis lupus) is a widely distributed top predator and ancestor of the domestic dog. To address questions about wolf relationships to each other and dogs, we assembled and analyzed a data set of 34 canine genomes. The divergence between New and Old World wolves is the earliest branching event and is followed by the divergence of Old World wolves and dogs, confirming that the dog was domesticated in the Old World. However, no single wolf population is more closely related to dogs, supporting the hypothesis that dogs were derived from an extinct wolf population. All extant wolves have a surprisingly recent common ancestry and experienced a dramatic population decline beginning at least ∼30 thousand years ago (kya). We suggest this crisis was related to the colonization of Eurasia by modern human hunter–gatherers, who competed with wolves for limited prey but also domesticated them, leading to a compensatory population expansion of dogs. We found extensive admixture between dogs and wolves, with up to 25% of Eurasian wolf genomes showing signs of dog ancestry. Dogs have influenced the recent history of wolves through admixture and vice versa, potentially enhancing adaptation. Simple scenarios of dog domestication are confounded by admixture, and studies that do not take admixture into account with specific demographic models are problematic.
The gray wolf (Canis lupus) is a dominant large predator that exerts important top-down effects on biodiversity (Levi and Wilmers 2012; Ripple et al. 2014). The species is widely distributed throughout the Holarctic (including the Nearctic and Palearctic regions), and as many as 32 subspecies have been described (Aggarwal et al. 2003). Gray wolves have an ancient origin, first appearing about 500 thousand years ago (kya) in Eurasia and in North America soon thereafter (Nowak 1979; Kurten and Anderson 1980). Initial studies based on mitochondrial DNA (mtDNA) data suggested that the gray wolf had a complex evolutionary history without clear worldwide phylogeographic structure (e.g., Wayne et al. 1992; Vilà et al. 1999). However, subsequent studies found subpopulation structure related to local environmental characteristics (e.g., Carmichael et al. 2001; Geffen et al. 2004; Pilot et al. 2006, 2010, 2014; Musiani et al. 2007; vonHoldt et al. 2011). Genome-wide approaches using SNP genotyping arrays have confirmed these environmentally related genetic partitions and demonstrated extensive admixture with coyotes and, to a more limited extent, with domestic dogs (Pilot et al. 2010, 2014; vonHoldt et al. 2010, 2011). Using complete genome sequence data of a wolf from Europe, Israel, and China, Freedman et al. (2014) found an unexpected recent coalescence of ∼30 kya, suggesting that wolves existing before that time were phylogenetically distinct, a result supported by genetic, isotopic, and morphologic analyses (Leonard et al. 2007; Thalmann et al. 2013). The wolves from these three regions also suffered a substantial bottleneck that initiated ∼15 kya, which was nearly coincident with the Wisconsin glacial maximum (Freedman et al. 2014). However, as inferred from genomic data, Zhang et al. (2014) found that Tibetan wolves experienced earlier and more dramatic population declines perhaps due to the extreme loss of wolf habitat with Late Pleistocene glaciations in the Tibetan Plateau. These findings suggest the recent worldwide history of wolves is complex and needs to be assessed with a fuller sample of genomes from throughout the historic range of the species.
The domestic dog (Canis lupus familiaris), a descendant of gray wolves, is the most widely abundant large carnivore (Vilà et al. 1999; Thalmann et al. 2013), but the specific region of origin is controversial. Previous genetic evidence suggested that dogs were domesticated either in the Middle East or East Asia (Savolainen et al. 2002; vonHoldt et al. 2010; Wang et al. 2013). However, a recent study based on ancient mtDNA analysis of dogs and wolves infers an origin in Europe from a now-extinct lineage of gray wolves (Thalmann et al. 2013). This result is consistent with whole-genome analysis, showing that none of the extant wolf lineages from putative domestication centers (Europe, Israel, and China) were more closely related to dogs (Freedman et al. 2014). Very recently, however, these conclusions were questioned by results from an extensive study of SNP genotypes in a worldwide sample of breed and village dogs, which concluded that dogs originated in Central Asia (Shannon et al. 2015). Consequently, we test for alternative regions of origin with a geographically broad sample of gray wolves.
The release of the boxer genome in 2005 (Lindblad-Toh et al. 2005) provided a high-quality dog reference for comparison to wolves and other canids (e.g., Wang et al. 2013; Freedman et al. 2014; Zhang et al. 2014; Koepfli et al. 2015). However, no studies have been performed to investigate population subdivision, demography, and relationships of gray wolves based on whole-genome sequences. In this study, we generate whole genomes of nine individual wolves, one coyote, and one golden jackal at 9–28× coverage using the Illumina HiSeq 2000 platform to geographically complement existing canine sequences. Combined with published genomes, we assemble a data set with 34 canid genomes to (1) assess relationship patterns across the entire geographic range of wolves; (2) affirm their recent demographic decline with a more geographically extensive sample; (3) assess admixture between dogs and wolves; and (4) explore the possibility of dog domestication outside the Middle East, Europe, and East Asia, which was not addressed in previous studies but is a possibility suggested by new findings (Shannon et al. 2015; Skoglund et al. 2015).
Results
Genome data and heterozygosity
In this study, we amassed the full genome sequences of 24 wolves, seven dogs (including the reference genome), and three outgroups (Fig. 1; Supplemental Table S1). Eleven of the individuals were uniquely sequenced in this study using the HiSeq 2000 platform with the remaining sequences obtained from previous studies (Wang et al. 2013; Freedman et al. 2014; Zhang et al. 2014; Supplemental Material).
Figure 1.

Sample distribution. Solid circles are samples sequenced in this study. Open circles indicate sequences from Zhang et al. (2014). Triangles and boxes indicate sequences from Wang et al. (2013) and Freedman et al. (2014), respectively. Species memberships are indicated by color: gray wolf (red), domestic dog (blue), coyote (green), and golden jackal (yellow). The reference dog genome is from a boxer.
To quantify genome-wide heterozygosity, we calculated the number of heterozygous SNPs over all sites (Fig. 2; Supplemental Table S2). The Mexican wolf had the lowest autosomal heterozygosity (0.00046), and the two Tibetan wolves also showed very low heterozygosity (0.0007 and 0.00086). Within European wolves, the Portuguese wolf showed the lowest heterozgosity (0.00101). However, the SNP rate was similar in all wolves (Supplemental Table S2). Within dogs, the basenji and dingo had the lowest heterozygosity (<0.001), with dingo having the lowest value of 0.00057 (Supplemental Table S2). We further calculated the heterozygosity of 5 Mb nonoverlapping windows across the 38 autosomal chromosomes (Supplemental Figs. S1–S4). The result confirmed that the low heterozygosity of two Tibetan wolves was evident across their entire genomes (Supplemental Fig. S1). The Mexican wolf exhibited extremely low heterozygosity in about half the chromosomes (Supplemental Fig. S4), and the Portuguese wolf also had very low heterozygosity in more than 15 autosomes (Supplemental Fig. S2). In contrast, the Inner Mongolia wolf 2 had very high heterozygosity across all chromosomes, even higher than other Inner Mongolia wolves, which partly may reflect lower genome coverage and a higher fraction of miscalled sites (Supplemental Figs. S1, S5). We also calculated the heterozygosity of exons and neutral regions (Supplemental Fig. S6).
Figure 2.

Total length of runs of homozygosity (ROHs) and heterozygosity. The black line is the total length of ROHs (Mb) in each genome, and the blue and red bars are the genome-wide heterozygosity with and without ROHs, respectively.
To avoid the effect of inbreeding in the calculation of heterozygosity, we removed runs of homozygosity (ROH, see below) and recalculated heterozygosity (Fig. 2). The results are similar to that of the full data set. For example, the inbred Mexican wolf still had the lowest heterozygosity within wolves, and two Tibetan wolves had higher values than the Mexican wolf but lower than other wolves.
Genome-wide phylogenetic tree and PCAs
Autosomal SNPs were used to construct a maximum likelihood (ML) tree (Fig. 3). The topology of ML trees with (Fig. 3) and without (Supplemental Fig. S7) the boxer reference genome is consistent with geographical proximity of populations and does not support any specific wolf population as more closely related and possibly ancestral to domestic dogs. Specifically, all the dogs are monophyletic and define a sister taxon with Eurasian gray wolves that excludes a role for New World wolves in dog origins and suggests that the divergence of the modern Eurasian wolf population is nearly coincident with that of domestic dogs. Among gray wolves, East Asian wolves form a single clade, whereas European and Middle Eastern wolves (including Indian wolf) form a separate grouping. The Middle Eastern wolf is aligned with European rather than Asian wolf sequences (Fig. 3). For the New World wolves, two Yellowstone wolves cluster together, and then the Mexican wolf is grouped with them, but the divergence is large, suggesting an ancient separation of the two populations. However, the long branch of the Mexican wolf lineage may reflect the effect of small historic population size as the species went extinct in the wild, which was compounded by an extreme founding bottleneck in the captive population (Fredrickson et al. 2007). Both demographic events would tend to inflate genetic distance values. Nonetheless, this finding supports a previous hypothesis that Mexican wolves represented an early migration into North America (Leonard et al. 2005).
Figure 3.

The maximum likelihood tree of 30 sequences. Numbers represent node support inferred from 100 bootstrap repetitions. The reference genome boxer was not included. The Israeli golden jackal is the outgroup.
Principal component analysis (PCA) with LD-pruned data excluding the three outgroups and four wolf sequences (Inner Mongolia wolf 2, Eastern Russian wolf, Yellowstone wolf 2, and Yellowstone wolf 3) showed that PC1 (20.2% of variation) divided the samples into three clusters: domestic dogs; highland Chinese wolves; and other gray wolves (Fig. 4A). Further, when two outlier highland wolves were removed (Tibet wolf 1 and Qinghai wolf 1), dogs were more tightly clustered and separated from all wolves on PC1, whereas PC2 distinguished high altitude wolves and the Central Russian wolf from all other wolves (27.7% of variation of both axes combined) (Fig. 4B). PC3 and PC4 of both data sets separate Old and New World wolves, with the Mexican wolf showing the greatest distinction (Fig. 4C,D). The results with only wolves and dogs excluded showed a similar pattern with regard to clusters of wolves (Supplemental Fig. S8). Critically, we found no support for a closer association of Chinese wolves with domestic dogs as suggested by previous studies (Savolainen et al. 2002; Pang et al. 2009; Ding et al. 2012; Wang et al. 2013). We also ran PCAs for different geographical regions (Supplemental Figs. S9–S11). These results are consistent with the tree-based analysis in Figure 3, but do not take into account rate variation between lineages that can bias inferences about the actual amount of divergence. Moreover, PCA is a graphical approach that highlights genetic clusters in the data and should not be used to infer genealogical relationships (Novembre and Stephens 2008).
Figure 4.

Principal component analyses. (A) PC1 and PC2 of dogs and 20 wolves; (B) PC1 and PC2 of dogs and 18 wolves, excluding the Tibetan wolf 1 and Qinghai wolf 1; (C) PC3 and PC4 of dogs and 20 wolves; (D) PC3 and PC4 of dogs and 18 wolves, excluding the Tibetan wolf 1 and Qinghai wolf 1. (□) Highland Asian wolves; (▵) lowland Asian wolves; (○) Middle Eastern wolves; (■) European wolves; (▲) dogs; (●) North American wolves.
PSMC
The pairwise sequentially Markovian coalescent model (PSMC) was applied to investigate the timing of population-specific demography. Here, we report only the results for the higher mutation rate (Fig. 5; Supplemental Fig. S12) to be consistent with Freedman et al. (2014), but consider the results from both rates (1.0 × 10−8 and 0.4 × 10−8 per generation) in the Discussion, as effective size and the timing of population size changes should differ by a factor of approximately 2.5 (Supplemental Figs. S13, S14). All the wolves exhibited similar demographic trajectories until ∼80 kya; thereafter, the four highland Chinese wolves showed very different trajectories when compared with all other wolves (Fig. 5A). The Tibetan wolves experienced a continuous population decline beginning ∼25 to 55 kya and did not experience further population growth; whereas the Qinghai wolf experienced population growth at the same time as the Tibetan wolf bottleneck (Fig. 5A). However, caution needs to be used in interpreting these results because they might be explained by ancestral population structure or reflect smoothing across time intervals (Freedman et al. 2014). Other wolves experienced population growth or stagnation from 25 to 55 kya, which overlaps the Greatest Lake Period (25–40 kya) (Li and Zhu 2001).
Figure 5.

Demographic history inferred using PSMC. Following Freedman et al. (2014) and Zhang et al. (2014), we used a generation time = 3 and a mutation rate = 1.0 × 10−8 per generation. The Tibetan wolf 1 and Inner Mongolia wolf 4 are shown in all the plots for comparison purposes. (A) All the Asian wolves; (B) all the European wolves, Middle Eastern wolves, and Indian wolf; (C) dogs; (D) Mexican wolf and Yellowstone wolves.
PSMC projections showed that the remaining wolves suffered a worldwide decline of two- to threefold beginning ∼8 to 25 kya (Fig. 5), which is associated with the end of the last glacial period (10.5–25 kya) and megafaunal extinctions. The Chinese wolves showed the most divergent trajectories with the Tibetan wolves, demonstrating a sharp decline beginning >25 kya and followed by a less precipitous decline in Qinghai wolves. In contrast, the lowland Chinese wolf populations do not initiate a decline until ∼10 kya (Fig. 5A). The Middle Eastern wolves (Israeli, Iranian, and Indian wolves) and European wolves exhibited slightly different demographic trajectories between 10 and 80 kya (Fig. 5B). All these wolves show evidence of a population decline beginning 25 kya. Domestic dogs had similar trajectories and experienced a population decline and demographic divergence from wolves beginning ∼50 kya (Fig. 5C). The three Yellowstone wolves had concordant trajectories (Fig. 5D). However, the Mexican wolf experienced a more severe bottleneck, which may reflect both a recent history of decline and demographic smoothing across the last 10,000-yr interval (see Hedrick et al. 1997; Freedman et al. 2014). The Israeli golden jackal had higher Ne than the Kenya golden jackal, and the California coyote exhibited a different trajectory as expected given its status as an independent lineage (Supplemental Fig. S12). In conclusion, a consistent result across all these trajectories is a decline in population sizes during the period of 8 to 25 kya, coincident with the expansion of modern humans worldwide and the development of technology for capturing large game (Van Valkenburgh et al. 2015).
Autozygosity segments
To assess the history of inbreeding, we quantified genome-wide ROH using PLINK (Fig. 2; Supplemental Fig. S15; Purcell et al. 2007). The Mexican wolf had the longest ROH with a total length of 1,569,600 kb (Fig. 2), which was consistent with a founding bottleneck and subsequent inbreeding (Hedrick et al. 1997; Fredrickson et al. 2007). In fact, the distribution of ROH in the Mexican wolf was distinct from that of all other wolves, and showed the highest fraction of autozygous long segments (Supplemental Fig. S15d), which suggests very recent inbreeding (e.g., Boyko et al. 2010). The two Tibetan wolves had the longest total length of ROH (947,844 kb and 835,018 kb) and the highest fraction of autozygous segments in Old World wolves, especially at small segment size. This result indicates ancient inbreeding in the Tibetan wolf population (Fig. 2; Supplemental Fig. S15a). The Italian wolf had the highest fraction of autozygous segments at smaller ROH sizes among European wolves, whereas the Portuguese wolf had more segments at longer sizes (Supplemental Fig. S15b). This contrasting pattern is consistent with previous genetic analysis, suggesting an ancient population decline in Italian wolves (Lucchini et al. 2004; Pilot et al. 2014) and historical records showing a very recent population decline in Portuguese wolves (Sastre et al. 2011). Within dogs, dingo and basenji had the greatest ROH (dingo: 1,097,810 kb; basenji: 589,502 kb). They also had a higher fraction of autozygous segments, especially in the size range <4 Mb than the Tibetan mastiff and three Chinese indigenous dogs (Fig. 2; Supplemental Fig. S15c), suggesting more ancient inbreeding perhaps in the founding population of dingo that arrived to Australia (>4 kya) and in the origin of the basenji, an ancient breed of domestic dog. These results show that novel demographic insights into population-specific demography are provided by PSMC and ROH analyses, which are consistent with known recent history and past environmental events.
ABBA-BABA
Multiple runs of the ABBA-BABA test were performed to assess gene flow between Old World wolves and dogs (Supplemental Table S3). The results showed that all the European wolves and the Israeli wolf had significant gene flow with basenji and boxer. For the Asian wolves, the two Russian wolves and all the lowland Chinese wolves had significant gene flow with all the Chinese indigenous dogs, Tibetan mastiff, and dingo, whereas the two Tibetan wolves did not show any significant admixture with any dogs (Supplemental Table S3). However, Qinghai wolf 1 showed significant gene flow with two of the three Chinese indigenous dogs, and both Qinghai wolves had gene flow with dingo. The Mexican wolf and Yellowstone wolf did not show any admixture signal with boxer, dingo, or Chinese indigenous dogs (Supplemental Table S3). We note that where admixture is detected from multiple dog samples in one or more wolf populations, it may suggest that gene flow actually occurred from the common ancestor of these dog into a specific wolf population or one that was ancestral to multiple wolf populations.
We estimated the proportion of Chinese indigenous dog ancestry in Asian wolves that had evidence for significant admixture and for which more than one dog defined the comparison pool (Supplemental Table S3, see above; Green et al. 2010; Durand et al. 2011). The proportion of Chinese indigenous dog ancestry in the two Russian wolves varied from 15.3% to 19.52%. The proportion of dog ancestry in the two Xinjiang wolves varied from 9.28% to 11.3%. The average proportion of the dog ancestry in four Inner Mongolia wolves was 10.86%, 12.06%, 13.16%, and 21.59% (Supplemental Table S4). These results suggest substantial dog ancestry in wolf populations worldwide, which is conceivable given the long coexistence of dogs and wild wolf populations (Thalmann et al. 2013; Freedman et al. 2014). The only Old World population not showing any dog ancestry is the Tibetan wolf, which is also the most divergent population in the PCA (Fig. 4), suggesting the dog component of wolf genomes may influence patterns of relationships. The high altitude wolf populations also have a very recent history of exposure to aboriginal dog populations considering that the area was only permanently colonized by humans ∼7 kya (Brantingham et al. 2010; Chen et al. 2015).
Regarding the European and Middle Eastern wolves, we used basenji and boxer to estimate the dog ancestry in these wolf genomes (Supplemental Table S4). The proportion of dog ancestry in Israeli wolf, Western Russian wolf, and Spanish wolf is >20%. Of the others, the Portuguese wolf had the smallest proportion at 7.97%, whereas the Croatian wolf had the largest at 13.76% dog ancestry (Supplemental Table S4). These findings indicate a highly variable but substantial dog ancestry in most all extant wolf populations.
Demographic inference with G-PhoCS
We used the Generalized Phylogenetic Coalescent Sampler method (G-PhoCS) to infer the demographic history of wolves and dogs, including ancestral population sizes, divergence times, and rates of gene flow (Fig. 6). The analysis shows that wolf populations diverged over a relatively short period of time from ∼11,000 to 13,000 yr ago (ya), assuming a per-generation mutation rate of μ = 1.0 × 10−8 and an average generation time of 3 yr (Fig. 6). If a slower mutation rate of μ = 0.4 × 10−8 is used as suggested by Skoglund et al. (2015), this period of time is increased by a factor of 2.5 to 27,500–32,500 ya (see Discussion). The divergence of New and Old World wolves is the oldest of these events at 12,500 ya, followed by divergence of Eastern and Western Eurasian wolves at 11,700 ya. The divergence times between sequences from Europe, the Middle East, and Asia fall within a relatively short period of time of ∼1600 yr. New World wolves show an intermediate divergence time of ∼5400 ya. We infer dogs diverged from wolves just before the Eurasian wolf population splits (11,700 ya; CI: 11,100–12,300 ya). This divergence time is only 285–1565 yr (95% CI) more recent than the divergence of New World wolves. The tree implies a considerable preancestry of extant dogs of 1400–2700 yr and a substantial level of divergence among existing dog lineages (dingo, Chinese indigenous dog, and basenji). In contrast, gray wolves have a much more recent common ancestry than expected from their fossil record, and all the population diverged over a narrow time period consistent with a bottleneck followed by a rapid population expansion across Eurasia.
Figure 6.
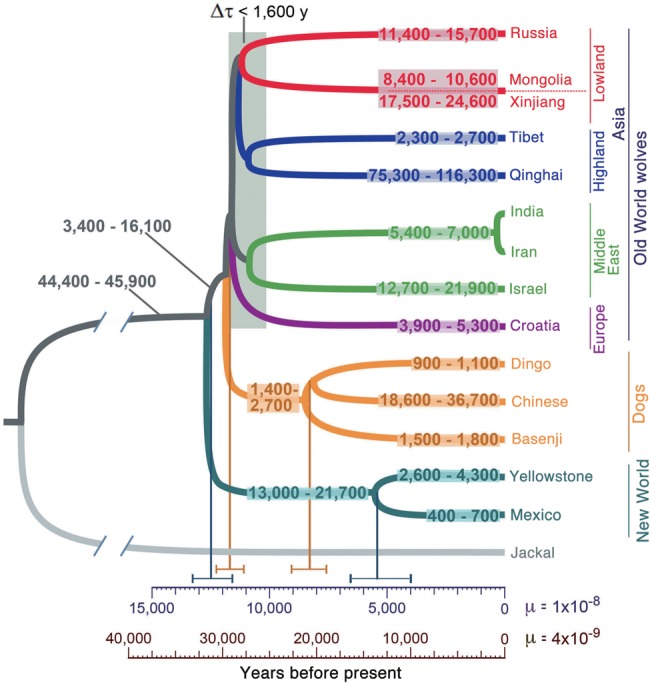
Demographic model inferred using G-PhoCS. Estimates of divergence times and effective population sizes (Ne) inferred by applying a Bayesian demography inference method (G-PhoCS) to sequence data from 13,647 putative neutral loci in a subset of 22 canid genomes (because of limitations in computational power). Estimates were obtained in four separate analyses (Methods; Supplemental Table 6). Ranges of Ne are shown and correspond to 95% Bayesian credible intervals. Estimates are calibrated by assuming a per-generation mutation rate of μ = 10−8. Mean estimates (vertical lines) and ranges corresponding to 95% Bayesian credible intervals are provided at select nodes. Scales are given in units of years by assuming an average generation time of 3 yr and two different mutation rates: μ = 10−8 (dark blue) and μ = 4 × 10−9 (brown). The model also considered gene flow between different population groups (see Table 1).
The population ancestral to Old and New World wolves was estimated to have a relatively large effective size of 45,100 individuals (CI: 44,400–45,900), when assuming a per-generation mutation rate of μ = 1.0 × 10−8. After divergence of Old and New World wolves, both populations experience decline, to 8000 individuals in Old World wolves and 17,300 individuals in New World wolves (Fig. 6; Supplemental Table S5). The Tibetan wolf had the smallest Ne within the Old World wolves (2500 individuals), whereas the lowland Chinese wolves had nearly fourfold larger Ne (Fig. 6; Supplemental Table S5). The ancestral population of New World wolves had a relatively large effective size of 17,300 individuals, implying a fairly modest bottleneck in the founding of the North American population. However, the two sampled populations have much lower inferred sizes of 3500 individuals for the Yellowstone wolf and 600 individuals for the Mexican wolf. The latter likely reflects a history of decline and extinction in the wild (see Discussion).
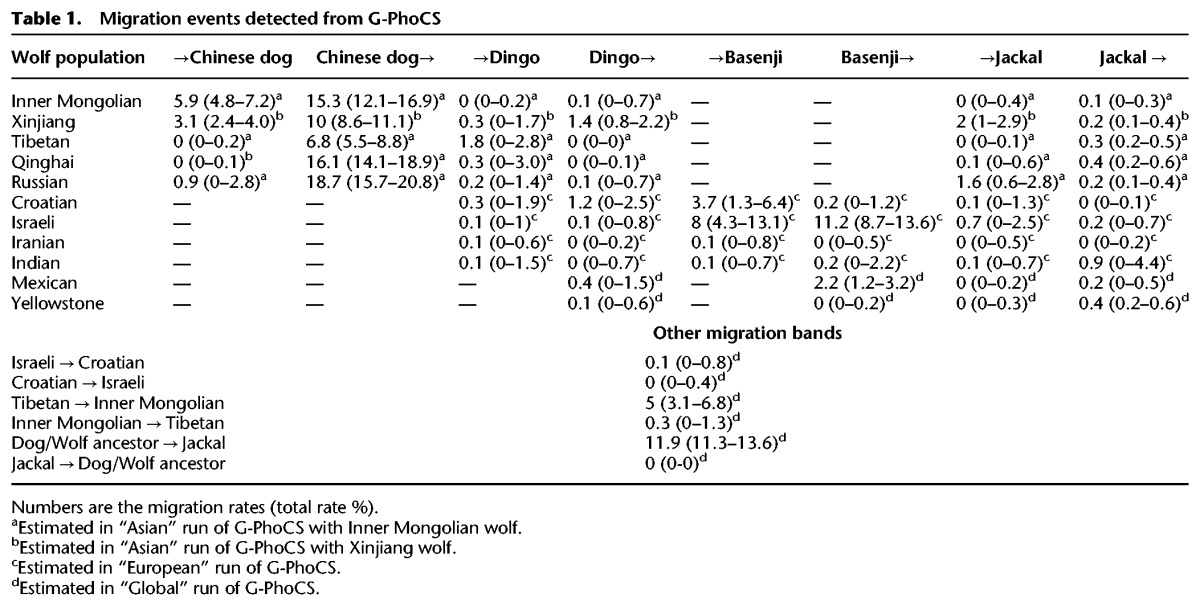
G-PhoCS models migration bands allowing a test of admixture from D-statistics. We infer relatively high rates of gene flow (5%–21%; aggregated 95% Bayesian credible intervals) from Chinese indigenous dogs to all Asian wolf populations and significant gene flow (2.4%–7.2%) in the opposite direction only for the lowland Chinese wolves (Table 1). Conversely, we find little evidence of admixture with the ancestors of the dingo (migration rates <3% in both directions). For the two highland Chinese wolf populations, G-PhoCS found relatively high rates of gene flow from Chinese indigenous dogs (Tibetan wolf: 5.5%–8.8%; Qinghai wolf: 14.1%–18.9%), although the ABBA-BABA test did not find evidence of admixture. In Western Eurasia, we observe high rates of gene flow between the Israeli wolf and basenji in both directions (4.3%–13.6%) and somewhat lower rates from Croatian wolf to basenji (1.3%–6.4%). Our findings of admixture between Chinese dogs and Asian wolves and Israeli wolf and basenji suggests admixture between the lineages ancestral to these breeds and wolf populations, as their geographic overlap is currently very limited. Consistent with previous results are the high rates of gene flow inferred from the population ancestral to all wolves and dogs into the golden jackal population (11.3%–13.6%) and much lower rates from several sampled wolf populations into the golden jackal population (up to 2.8%) (Freedman et al. 2014). The higher observed value of admixture between golden jackal and the common ancestor of modern wolves suggests an ancient admixture event. Finally, we infer low, but significant, levels of gene flow from the basenji into the Mexican wolf population (1.2%–3.2%), suggesting that like other wolf populations, the New World wolves also experienced admixture with dogs or share a common ancestor with Old World wolves that experienced admixture.
Table 1.
Migration events detected from G-PhoCS
Discussion
Genetic diversity and relationships of Old and New World wolves
Analysis of complete genome sequence data adds considerable resolution to the evolutionary relationships of gray wolves and domestic dogs. First, the genome-wide phylogenetic tree shows that the earliest split was between New and Old World wolves, which was followed by divergence between Old World wolves and dogs (Fig. 3). This result confirms dogs were domesticated in the Old World. In addition, the finding that no single wolf population is more closely clustered with domestic dogs supports the hypothesis that dogs were derived from a now extinct population of Late Pleistocene wolf (Thalmann et al. 2013; Freedman et al. 2014). However, the divergence time suggested by G-PhoCS (11,700 ya; CI: 11,100–12,300 ya) is more recent than estimates based on ancient DNA analysis of early dogs and wolves (27,000 ya) (Thalmann et al. 2013). These differences might be caused by inflated mutation rates in the neutral regions used in this study, undetected admixture with dogs, or other assumptions of the underlying G-PhoCS model. The existence of dog fossils older than this recent divergence date, and confirmed by mtDNA sequence data, supports a more ancient origination (Thalmann et al. 2013; Skoglund et al. 2015). In fact, if the mutation rates associated with Skoglund et al. (2015) are used, the divergence time increases to ∼29 kya, a value close to their estimate of 27 kya (Fig. 6). Finally, within the Old World clade, wolf and dog represent sister taxa. Therefore, suggestions that the dog or dingo are a separate species (Canis familiaris) (e.g., Crowther et al. 2014) would cause gray wolves to be a polyphetic taxon; and consequently, our results support dogs as a divergent subspecies of the wolf. This result has societal significance as legislation in some countries and regional governments consider wolves and dogs as distinct species restricting the possession, interbreeding, or the use of vaccines and medications in wolves or dog–wolf hybrids if they have only been approved for use in dogs. In this sense, analysis of evolutionary history informs law and veterinary practice, as dog lineages are nearly as distinct from one another as wolves are from dogs, and the justification for treating dogs and wolves differently is questionable.
The evolutionary tree (Fig. 3) and PCA (Fig. 4) show that the Mexican wolf is a divergent form of gray wolf, suggesting it is a remnant of an early invasion into North America (García-Moreno et al. 1996; Leonard et al. 2005; vonHoldt et al. 2011) and contradicting suggestions that it is not a distinct subspecies (Cronin et al. 2015). The ROH and genome-wide heterozygosity results (Fig. 2) also showed that the Mexican wolf is a highly inbred population (vonHoldt et al. 2011). The subspecies had the smallest effective population size of only 600 individuals in the sampled wolf populations (Fig. 6; Supplemental Table S5). Further, the high long-range ROH in the Mexican wolf implies a long-term decline, followed by a small founding population and inbreeding in the captive population (Hedrick et al. 1997; Fredrickson et al. 2007). These results justify immediate conservation actions to protect this endangered and distinct wolf lineage. Further, population numbers should be increased through captive breeding and in situ conservation to prevent additional genetic erosion. Currently, such efforts have been hindered by the lack of an informed management plan (Wayne and Hedrick 2011; Hendricks et al. 2016).
The Tibetan wolf was found to be the most highly divergent Old World wolf, given its distinct position in the phylogenetic tree (Fig. 3) and the PCA plot (Fig. 4). It also exhibited extremely low heterozygosity (Fig. 2; Supplemental Figs. S1–S4), suggesting that it experienced a historical bottleneck, and only recently recolonized much of the Tibetan Plateau. Indeed, PSMC revealed that the Tibetan wolf suffered a substantial population bottleneck that began ∼55 kya (mutation rate 1.0 × 10−8) or >100 kya, assuming a slower mutation rate, and then declined to the present day (Fig. 5A). Notably, all other wolves showed evidence of growth during the Greatest Lake Period from ∼25 to 55 kya (Fig. 5). The severe habitat loss during glaciations probably contributed to the dramatic population decline of the Tibetan wolf between 10 and 55 kya (Xu and Shen 1995; Yi et al. 2005; Clark et al. 2009; Chevalier et al. 2011; Heyman 2014; Zhang et al. 2014). In addition, both archaeological and genetic analysis suggest that the first colonization might be as early as 30 kya (Aldenderfer 2011), and little evidence exists for permanent human occupation before 7 kya (Brantingham et al. 2010; Chen et al. 2015). Therefore, the appearance of human settlements may have contributed to the decline of Tibetan wolf population but did not initiate the population bottleneck more than 50 kya. Finally, Tibetan wolves had the longest total length of ROHs of the Old World wolves (Fig. 2), and a large proportion of their ROHs are in relatively short segments (Supplemental Fig. S15), which suggests that it experienced ancient inbreeding. Moreover, the ABBA-BABA test did not detect substantial gene flow between Tibetan wolf and dogs, suggesting dog admixture did not contribute to ROH (see discussion below). In summary, we suggest that the unique high altitude environment and history of the Tibetan Plateau made wolves there more susceptible to habitat loss, genetic isolation, and allowed for local adaptation. Consequently, these conditions resulted in the evolution of the most distinct wolf population in the Old World.
Geographical structure is evident within Old World wolves (Fig. 3). Previously, analysis of Eurasian wolves with mtDNA control region sequences did not reveal any distinct genetic partitions and suggested modern wolves originated over 250 kya (Vilà et al. 1999). However, European and Middle Eastern partitions were apparent in genome-wide SNP data (vonHoldt et al. 2011). The one-million-year divergence time between wolves and coyotes used previously was based on fossil occurrence data; and given the dynamics of morphological turnover in the coyote lineage (Meachen and Samuels 2012), first occurrence of coyote-like specimens may not accurately reveal the ancestry of modern forms. Our results and those from previous studies (Freedman et al. 2014; Koepfli et al. 2015; Skoglund et al. 2015) suggest the one-million-year divergence time may be inflated by a factor of 20 or more, and modern Eurasian wolves coalesce ∼13 kya or ∼32.5 kya, the latter using the slower mutation rate from Skoglund et al. (2015). Importantly, the slower rate leads to divergence dates more consistent with the presence of ancient dog fossils well before 15,000 yr ago. Nonetheless, the Skoglund et al. rate needs additional confirmation because it is based on a single fossil specimen with only onefold sequencing depth and used only a subset of DNA sequence to calculate the rate (Skoglund et al. 2015). Thus, until more direct measurements of mutation rates become available, fossil calibration will remain the main source of uncertainty in the timing of key events in canid evolutionary history.
The PSMC results revealed that all wolves shared a similar trajectory before ∼100–125 kya (mutation rate 0.4 × 10−8) or ∼30–50 kya (mutation rate 1.0 × 10−8). In combination, the dating and PSMC results suggested that over the last million years, numerous wolf-like forms existed but that turnover was high, and modern wolves were not the lineal ancestors of dogs (Leonard et al. 2005; Thalmann et al. 2013; Freedman et al. 2014). Indeed, the population size of the Croatian wolf reduced about 10-fold compared to the wolf ancestor, and Yellowstone wolf and Mexican wolf also reduced five- to 28-fold (Fig. 6; Supplemental Table S5). This pattern of population reduction and turnover also is supported by recent mtDNA sequence analysis of modern and ancient wolves from the Last Glacial Maximum (Leonard et al. 2007; Pilot et al. 2010; Thalmann et al. 2013) and the dynamic pattern of turnover in other large carnivores such as brown and polar bears, hyenas, and lions as inferred from genetic data (Miller et al. 2012; Cho et al. 2013).
Finally, even assuming a slower mutation rate, our results imply a remarkably recent coalescence of extant wolves several hundred thousand years after the appearance of wolf-like canids (Wayne and Ostrander 2007). Both slow and fast mutation rate estimates are consistent with the possibility that modern humans impacted the demography of gray wolves as they colonized Eurasia, encountered wolves, domesticated some, and possibly caused the decline of others. Humans are the most effective competitor of large carnivores and could have readily removed them from ecosystems as they do today. Additionally, the presence of large domestic dogs may have accelerated the rate of decline of carnivores that competed with humans (e.g., Shipman 2015). Our results imply that the effect of humans on large predators may have preceded the megafaunal extinctions ∼10 kya and may represent one of the earliest anthropogenic causes of decline in animal populations.
Admixture and relationships to domestic dogs
None of our wolf sequences cluster exclusively with domestic dogs, supporting the hypothesis based on only three wolf genomes (Freedman et al. 2014) and ancient DNA (Thalmann et al. 2013) that the immediate gray wolf ancestor of dogs is now extinct. Nonetheless, modern gray wolves have likely influenced the recent history of domestic dogs through admixture. Both the ABBA-BABA tests and G-PhoCS support the notion of extensive admixture between dogs and wolves, with up to 20% of the genome of East Asian wolves showing signs of dog ancestry. We also detected that the genomes of European and Middle Eastern wolves had ∼7%–25% dog ancestry. Most of the observed gene flow events have not been reported previously. Interestingly, the two highland wolf populations of the Tibetan Plateau showed no evidence of admixture in the ABBA-BABA tests, but G-PhoCS did infer elevated migration rates from Chinese indigenous dogs into these populations (Tibetan wolf: 5.5%–8.8%; Qinghai wolf: 14.1%–18.9%). Conceivably, this finding may be a result of gene flow from dogs into the population ancestral to all modern wolves, which influenced the distribution of coalescent times but cannot be detected using D-statistics because it similarly affected all wolf populations.
For comparison, using ABBA-BABA tests, it was found that modern humans admixed with Neanderthals over 40 kya, but no more than 5% of the modern human genome could be attributed to admixture (Green et al. 2010), suggesting that wolves and dogs have more extensive and regionally based admixture. As in Neanderthals, admixture may have enhanced adaptation in wolves. For example, admixture of pre-Columbian dogs and wolves in North America transferred the black coat color locus to wolves, conferring greater longevity and resulting in a continent-wide selective sweep (Anderson et al. 2009; Coulson et al. 2011). However, in the North American wolves sampled, no apparent trace of admixture remains elsewhere in the genome (Anderson et al. 2009). The persistent admixture between dogs and wolves suggests a unique mode of evolution in which mutations that occur independently in dogs and wolves, under dramatically different selective regimes, can be shared and potentially accelerate the process of evolution. Such coupled evolutionary histories may exist in other vertebrate species as well, such as in wild and domestic pigs and in brown bears and polar bears (Groenen et al. 2012; Miller et al. 2012).
Finally, admixture can have a confounding effect on inferences about dog domestication history. Specifically, past inferences about dog origins based on private SNPs shared with dogs (vonHoldt et al. 2010), greater genome-wide similarity between Chinese wolves and dogs (Wang et al. 2013), or lower LD (Shannon et al. 2015) may reflect regional admixture with wolves and gene flow among dog populations rather than the geographic origin of domestication. Similarly, highly divergent breeds may have more admixture and wolf ancestry retained in their genome. Potentially, this bias might be removed by applying analytical approaches that excise dog segments from wolf genomes. However, direct tracking of genetic changes in wolves and dogs through ancient DNA analysis may be a more robust approach (Grimm 2015).
Methods
Samples and sequencing
We sequenced genomes of dogs, wolves, and other wild canids from Africa, Asia, Europe, the Middle East, and North America. Together with published canid genomes, we generated a final data set with 34 full genome sequences at 9–28× coverage with an average coverage of 29.8× (Fig. 1; see Supplemental Material).
Mapping short reads and genotyping
The 100-bp pair-end (PE) short reads of each sample were aligned to the dog genome (CanFam3.1) using Bowtie 2 (Langmead and Salzberg 2012) under the local alignment algorithm with very sensitive model and proper insert sizes of each sample. Default options were used for other parameters. Then, we applied Picard and GATK toolsets (DePristo et al. 2011) to process the alignments to SNP calls. The whole pipeline converted the short reads to BAM format alignment files, and then generated genotype calls in Variant Call Format (VCF). The pipeline is the same as used in our previous studies (Fan et al. 2014; Freedman et al. 2014; Zhang et al. 2014). We applied a series of data quality filters to improve the quality of genotype calls (see Supplemental Material).
Phylogenetic tree and PCA
A ML tree from whole-genome SNP data was constructed using SNPhylo (Lee et al. 2014). SNPhylo transforms genotype data into a structured data array (Bioconductor gdsfmt) and then generates and aligns SNP sequences and constructs the phylogenetic trees. The program was run with 100 bootstrap repetitions, and only one outgroup was used (Israeli golden jackal) due to the software's internal limitations.
PCA was performed using the pairwise allele-sharing genetic distance. Following vonHoldt et al. (2011), sites exhibiting apparent strong local linkage disequilibrium (R2 > 0.5) were filtered using the --indep option in PLINK (--indep 50 5 0.2) (Purcell et al. 2007). To improve resolution among wolves, the two golden jackals and coyote were removed from the PCA because they were too divergent from dogs and wolves, and their inclusion compressed the scatter among wolves on the first few PCs. The lower coverage genomes (<10-fold; Inner Mongolia wolf 2, Eastern Russian wolf, and Yellowstone wolf 3) were also removed due to their potential high genotype error (Supplemental Fig. S5). Additional PCA was also performed excluding one Tibetan wolf and one Qinghai wolf based on the observation that highland Chinese wolves were similar to one another but were highly divergent from all other wolves (Zhang et al. 2014). Finally, additional PCAs were performed with samples from specific geographic regions, such as Asia or Europe, and including only gray wolves. In both tree analyses and PCAs, we excluded the Yellowstone wolf 2 because it is the offspring of Yellowstone wolf 1 (mother) and Yellowstone wolf 3 (father).
Inference of population size changes through time with PSMC
We used PSMC (Li and Durbin 2011) to infer demographic history. The following parameters were used: numbers of iterations = 25; time interval = 64 × 1; and generation time = 3 (Freedman et al. 2014; Zhang et al. 2014). Our previous studies used a mutation rate of 1.0 × 10−8 per generation, a commonly applied value. However, one recent study based on ancient wolf genome sequences estimated that the mutation rate was only 0.4 × 10−8 per generation (Skoglund et al. 2015). Therefore, we used both mutation rates in our study to bracket estimates of divergence time and effective population size.
Runs of homozygosity analysis
ROHs were calculated with PLINK (Purcell et al. 2007). The input data here is the same data in PCA. We then searched for ROHs spanning at least 500 homozygous SNPs in 1 Mb nonoverlapping windows, allowing for a maximum of five heterozygous sites and 30 missing genotypes per window.
Detection of gene flow using the D-statistic
To test whether there was gene flow between wolves and dogs in each geographical region after their divergence, we applied the ABBA-BABA test (D-statistic) between closely related populations by detecting differences in allele sharing between two lineages (P1 and P2) with a third lineage (P3) (Durand et al. 2011; see Supplemental Material). Under the assumption of one gene flow event that is recent compared to the divergence of dogs and wolves, we further used the Durand et al. (2011) equation to estimate the proportion of dog ancestry in the wolf genomes (see Supplemental Material).
Demographic inference with G-PhoCS
The G-PhoCS method (Gronau et al. 2011) was used to estimate divergence times, population sizes, and migration rates. Considering the computational resources required, our analysis focused on a subset of 22 of the 33 genomes that represent all 11 wolf populations, four dog populations, and the Israeli golden jackal as outgroup (Supplemental Table S6). Alignments of the 22 genomes were done over the 13,647 neutral loci designed by Freedman et al. (2014) for use in demographic inference. We ran several analyses (see Supplemental Material) using the standard settings and assumed standard priors for model parameters, as described by Gronau et al. (2011).
Data access
The data generated from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under accession number SRP044399.
Supplementary Material
Acknowledgments
This work was supported by National Science Foundation (NSF) grant EF-1021397 (R.K.W., R.M.S.), the National Key Technology R&D Program of China 2012BAC01B06 (Z.F., B.S.Y.), ICREA, EMBO YIP 2013 and MICINN BFU2014-55090-P (T.M.B.), National Human Genome Research Institute (NHGRI) grant R00HG005846 (J.X.), UC MEXUS-CONACYT doctoral fellowship 213627 (D.O.D.V.), the Chengdu Giant Panda Breeding Research Foundation CPF-yan-2012-10 (W.Z., Z.Z.), and the grant PRIC from Fundació Barcelona Zoo and Ajuntament de Barcelona (O.R.). We thank Prof. Jeffrey Brantingham at the Department of Anthropology, UCLA, and Dr. Xiaoming Wang at the Natural History Museum of Los Angeles County, Los Angeles, for valuable discussions on the Tibetan Plateau.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.197517.115.
References
- Aggarwal RK, Ramadevi J, Singh L. 2003. Ancient origin and evolution of the Indian wolf: evidence from mitochondrial DNA typing of wolves from Trans-Himalayan region and Pennisular India. Genome Biol 4: P6. [Google Scholar]
- Aldenderfer M. 2011. Peopling the Tibetan plateau: insights from archaeology. High Alt Med Biol 12: 141–147. [DOI] [PubMed] [Google Scholar]
- Anderson TM, vonHoldt BM, Candille SI, Musiani M, Greco C, Stahler DR, Smith DW, Padhukasahasram B, Randi E, Leonard JA, et al. 2009. Molecular and evolutionary history of melanism in North American gray wolves. Science 323: 1339–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyko AR, Quignon P, Li L, Schoenebeck J, Degenhardt JD, Lohmueller KE, Zhao K, Brisbin A, Parker HG, vonHoldt BM, et al. 2010. A simple genetic architecture underlies morphological variation in dogs. PLoS Biol 8: e1000451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantingham PJ, Rhode D, Madsen DB. 2010. Archaeology augments Tibet's genetic history. Science 329: 1467. [DOI] [PubMed] [Google Scholar]
- Carmichael LE, Nagy JA, Larter NC, Strobeck C. 2001. Prey specialization may influence patterns of gene flow in wolves of the Canadian Northwest. Mol Ecol 10: 2787–2798. [DOI] [PubMed] [Google Scholar]
- Chen FH, Dong GH, Zhang DJ, Liu XY, Jia X, An CB, Ma MM, Xie YW, Barton L, Ren XY, et al. 2015. Agriculture facilitated permanent human occupation of the Tibetan Plateau after 3600 B.P. Science 347: 248–250. [DOI] [PubMed] [Google Scholar]
- Chevalier ML, Hilley G, Tapponnier P, Van der Woerd J, Jing LZ, Finkel RC, Ryerson FJ, Li HB, Liu XH. 2011. Constraints on the late Quaternary glaciations in Tibet from cosmogenic exposure ages of moraine surfaces. Quat Sci Rev 30: 528–554. [Google Scholar]
- Cho YS, Hu L, Hou H, Lee H, Xu J, Kwon S, Oh S, Kim HM, Jho S, Kim S, et al. 2013. The tiger genome and comparative analysis with lion and snow leopard genomes. Nat Commun 4: 2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark PU, Dyke AS, Shakun JD, Carlson AE, Clark J, Wohlfarth B, Mitrovica JX, Hostetler SW, McCabe AM. 2009. The last glacial maximum. Science 325: 710–714. [DOI] [PubMed] [Google Scholar]
- Coulson T, MacNulty DR, Stahler DR, vonHoldt B, Wayne RK, Smith DW. 2011. Modeling effects of environmental change on wolf population dynamics, trait evolution, and life history. Science 334: 1275–1278. [DOI] [PubMed] [Google Scholar]
- Cronin MA, Cánovas A, Bannasch DL, Oberbauer AM, Medrano JF. 2015. Wolf subspecies: reply to Weckworth, et al. and Fredrickson, et al. J Hered 106: 417–419. [DOI] [PubMed] [Google Scholar]
- Crowther MS, Fillios M, Colman N, Letnic M. 2014. An updated description of the Australian dingo (Canis dingo Meyer, 1793). J Zool 293: 192–203. [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding ZL, Oskarsson M, Ardalan A, Angleby H, Dahlgren LG, Tepeli C, Kirkness E, Savolainen P, Zhang YP. 2012. Origins of domestic dog in southern East Asia is supported by analysis of Y-chromosome DNA. Heredity 108: 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand EY, Patterson N, Reich D, Slatkin M. 2011. Testing for ancient admixture between closely related populations. Mol Biol Evol 28: 2239–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Zhao G, Li P, Osada N, Xing J, Yi Y, Du L, Silva P, Wang H, Sakate R, et al. 2014. Whole genome sequencing of Tibetan macaque (Macaca thibetana) provides new insight into the macaque evolutionary history. Mol Biol Evol 31: 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredrickson RJ, Siminski P, Woolf M, Hedrick PW. 2007. Genetic rescue and inbreeding depression in Mexican wolves. Proc Biol Sci 274: 2365–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman AH, Gronau I, Schweizer RM, Ortega-Del Vecchyo D, Han E, Silva PM, Galaverni M, Fan Z, Marx P, Lorente-Galdos B, et al. 2014. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet 10: e1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Moreno J, Matocq MD, Roy MS, Geffen E, Wayne RK. 1996. Relationships and genetic purity of the endangered Mexican wolf based on analysis of microsatellite loci. Conserv Biol 10: 376–389. [Google Scholar]
- Geffen E, Anderson MJ, Wayne RK. 2004. Climate and habitat barriers to dispersal in the highly mobile grey wolf. Mol Ecol 13: 2481–2490. [DOI] [PubMed] [Google Scholar]
- Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH, et al. 2010. A draft sequence of the Neandertal genome. Science 328: 710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D. 2015. How the wolf became the dog. Science 348: 277. [DOI] [PubMed] [Google Scholar]
- Groenen MA, Archibald AL, Uenishi H, Tuggle CK, Takeuchi Y, Rothschild MF, Rogel-Gaillard C, Park C, Milan D, Megens HJ, et al. 2012. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491: 393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronau I, Hubisz MJ, Gulko B, Danko CG, Siepel A. 2011. Bayesian inference of ancient human demography from individual genome sequences. Nat Genet 43: 1031–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW, Miller PS, Geffen E, Wayne RK. 1997. Genetic evaluation of the three captive Mexican wolf lineages. Zoo Biology 16: 47–69. [Google Scholar]
- Hendricks SA, Clee PR, Harrigan RJ, Pollinger JP, Freedman AH, Callas R, Figura PJ, Wayne RK. 2016. Re-defining historical geographic range in species with sparse records: implications for the Mexican wolf reintroduction program. Biol Conserv 194: 48–57. [Google Scholar]
- Heyman J. 2014. Paleoglaciation of the Tibetan Plateau and surrounding mountains based on exposure ages and ELA depression estimates. Quat Sci Rev 91: 30–41. [Google Scholar]
- Koepfli KP, Pollinger J, Godinho R, Robinson J, Lea A, Hendricks S, Schweizer RM, Thalmann O, Silva P, Fan Z, et al. 2015. Genome-wide evidence reveals that African and Eurasian golden jackals are distinct species. Curr Biol 25: 2158–2165. [DOI] [PubMed] [Google Scholar]
- Kurten B, Anderson E. 1980. Pleistocene mammals of North America. Columbia University Press, New York. [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH, Guo H, Wang X, Kim C, Paterson AH. 2014. SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genomics 15: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JA, Vilà C, Wayne RK. 2005. Legacy lost: genetic variability and population size of extirpated US grey wolves (Canis lupus). Mol Ecol 14: 9–17. [DOI] [PubMed] [Google Scholar]
- Leonard JA, Vilà C, Fox-Dobbs K, Koch PL, Wayne RK, Van Valkenburgh B. 2007. Megafaunal extinctions and the disappearance of a specialized wolf ecomorph. Curr Biol 17: 1146–1150. [DOI] [PubMed] [Google Scholar]
- Levi T, Wilmers CC. 2012. Wolves–coyotes–foxes: a cascade among carnivores. Ecology 93: 921–929. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2011. Inference of human population history from individual whole-genome sequences. Nature 475: 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Zhu Li. 2001. “Greatest lake period” and its palaeo-environment on the Tibetan Plateau. J Geogr Sci 11: 34–42. [Google Scholar]
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ III, Zody MC, et al. 2005. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438: 803–819. [DOI] [PubMed] [Google Scholar]
- Lucchini V, Galov A, Randi E. 2004. Evidence of genetic distinction and long-term population decline in wolves (Canis lupus) in the Italian Apennines. Mol Ecol 13: 523–536. [DOI] [PubMed] [Google Scholar]
- Meachen JA, Samuels JX. 2012. Evolution in coyotes (Canis latrans) in response to the megafaunal extinctions. Proc Natl Acad Sci 109: 4191–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller W, Schuster SC, Welch AJ, Ratan A, Bedoya-Reina OC, Zhao F, Kim HL, Burhans RC, Drautz DI, Wittekindt NE, et al. 2012. Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proc Natl Acad Sci 109: E2382–E2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiani M, Leonard JA, Cluff HD, Gates CC, Mariani S, Paquet PC, Vilà C, Wayne RK. 2007. Differentiation of tundra/taiga and boreal coniferous forest wolves: genetics, coat colour and association with migratory caribou. Mol Ecol 16: 4149–4170. [DOI] [PubMed] [Google Scholar]
- Novembre J, Stephens M. 2008. Interpreting principal component analyses of spatial population genetic variation. Nat Genet 40: 646–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak RM. 1979. North American Quaternary Canis. Monograph of the Museum of Natural History. University of Kansas, Lawrence, KS. [Google Scholar]
- Pang JF, Kluetsch C, Zou XJ, Zhang AB, Luo LY, Angleby H, Ardalan A, Ekström C, Sköllermo A, Lundeberg J, et al. 2009. mtDNA data indicate a single origin for dogs south of Yangtze River, less than 16,300 years ago, from numerous wolves. Mol Biol Evol 26: 2849–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilot M, Jedrzejewski W, Branicki W, Sidorovich VE, Jedrzejewska B, Stachura K, Funk SM. 2006. Ecological factors influence population genetic structure of European grey wolves. Mol Ecol 15: 4533–4553. [DOI] [PubMed] [Google Scholar]
- Pilot M, Branicki W, Jędrzejewski W, Goszczyński J, Jędrzejewska B, Dykyy I, Shkvyrya M, Tsingarska E. 2010. Phylogeographic history of grey wolves in Europe. BMC Evol Biol 10: 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilot M, Greco C, vonHoldt BM, Jędrzejewska B, Randi E, Jędrzejewski W, Sidorovich VE, Ostrander EA, Wayne RK. 2014. Genome-wide signatures of population bottlenecks and diversifying selection in European wolves. Heredity 112: 428–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripple WJ, Estes JA, Beschta RL, Wilmers CC, Ritchie EG, Hebblewhite M, Berger J, Elmhagen B, Letnic M, Nelson MP, et al. 2014. Status and ecological effects of the world's largest carnivores. Science 343: 1241484. [DOI] [PubMed] [Google Scholar]
- Sastre N, Vilà C, Salinas M, Bologov VV, Urios V, Sánchez A, Francino O, Ramírez O. 2011. Signatures of demographic bottlenecks in European wolf populations. Conserv Genet 12: 701–712. [Google Scholar]
- Savolainen P, Zhang YP, Luo J, Lundeberg J, Leitner T. 2002. Genetic evidence for an East Asian origin of domestic dogs. Science 298: 1610–1613. [DOI] [PubMed] [Google Scholar]
- Shannon LM, Boyko RH, Castelhano M, Corey E, Hayward JJ, McLean C, White ME, Abi Said M, Anita BA, Bondjengo NI, et al. 2015. Genetic structure in village dogs reveals a Central Asian domestication origin. Proc Natl Acad Sci 112: 13639–13644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipman P. 2015. The invaders: how humans and their dogs drove Neanderthals to extinction. Belknap Press of Harvard University Press, Cambridge, MA. [Google Scholar]
- Skoglund P, Ersmark E, Palkopoulou E, Dalén L. 2015. Ancient wolf genome reveals an early divergence of domestic dog ancestors and admixture into high-latitude breeds. Curr Biol 25: 1515–1519. [DOI] [PubMed] [Google Scholar]
- Thalmann O, Shapiro B, Cui P, Schuenemann VJ, Sawyer SK, Greenfield DL, Germonpré MB, Sablin MV, López-Giráldez F, Domingo-Roura X, et al. 2013. Complete mitochondrial genomes of ancient canids suggest a European origin of domestic dogs. Science 342: 871–874. [DOI] [PubMed] [Google Scholar]
- Van Valkenburgh B, Hayward MW, Ripple WJ, Meloro C, Roth VL. 2015. The impact of large terrestrial carnivores on Pleistocene ecosystems. Proc Natl Acad Sci. 10.1073/pnas.1502554112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilà C, Amorim IR, Leonard JA, Posada D, Castroviejo J, Petrucci-Fonseca F, Crandall KA, Ellegren H, Wayne RK. 1999. Mitochondrial DNA phylogeography and population history of the grey wolf Canis lupus. Mol Ecol 8: 2089–2103. [DOI] [PubMed] [Google Scholar]
- vonHoldt BM, Pollinger JP, Lohmueller KE, Han E, Parker HG, Quignon P, Degenhardt JD, Boyko AR, Earl DA, Auton A, et al. 2010. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 464: 898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vonHoldt BM, Pollinger JP, Earl DA, Knowles JC, Boyko AR, Parker H, Geffen E, Pilot M, Jedrzejewski W, Jedrzejewska B, et al. 2011. A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res 12: 1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GD, Zhai W, Yang HC, Fan RX, Cao X, Zhong L, Wang L, Liu F, Wu H, Cheng LG, et al. 2013. The genomics of selection in dogs and the parallel evolution between dogs and humans. Nat Commun 4: 1860. [DOI] [PubMed] [Google Scholar]
- Wayne RK, Hedrick P. 2011. Genetics and wolf conservation in the American West: lessons and challenges. Heredity 107: 16–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne RK, Ostrander EA. 2007. Lessons learned from the dog genome. Trends Genet 23: 557–567. [DOI] [PubMed] [Google Scholar]
- Wayne RK, Lehman N, Allard MW, Honeycutt RL. 1992. Mitochondrial DNA variability of the gray wolf: genetic consequences of population decline and habitat fragmentation. Conserv Biol 6: 559–569. [Google Scholar]
- Xu DM, Shen YP. 1995. On ancient ice-sheet and ice age in the Tibetan plateau. J Glaciol Geocryol 17: 213–229. [Google Scholar]
- Yi CL, Cui ZJ, Xiong HG. 2005. Numerical period of Quaternary glaciations in China. Quatern Sci 25: 609–611. [Google Scholar]
- Zhang W, Fan Z, Han E, Hou R, Zhang L, Galaverni M, Huang J, Liu H, Silva P, Li P, et al. 2014. Hypoxia adaptations in the grey wolf (Canis lupus chanco) from Qinghai-Tibet Plateau. PLoS Genet 10: e1004466. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.