

Abstract
Acute myocardial infarction (AMI) and sudden cardiac death (SCD) are among the most serious and catastrophic of acute cardiac disorders, accounting for hundreds of thousands of deaths each year worldwide. Although the incidence of AMI has been decreasing in the US according to the American Heart Association, heart disease is still the leading cause of mortality in adults. In most cases of AMI and in a majority of cases of SCD, the underlying pathology is acute intraluminal coronary thrombus formation within an epicardial coronary artery leading to total or near-total acute coronary occlusion. This article summarizes our current understanding of the pathophysiology of these acute coronary syndromes and briefly discusses new approaches currently being researched in an attempt to define and ultimately reduce their incidence.
Introduction
AMI and SCD are among the most serious and catastrophic of acute cardiac disorders, accounting for hundreds of thousands of deaths each year worldwide. Although the incidence of AMI has been decreasing in the US according to the American Heart Association, heart disease is still the leading cause of mortality in adults [1]. In most cases of AMI and in a majority of cases of SCD, the underlying pathology is acute intraluminal coronary thrombus formation within an epicardial coronary artery leading to total or near total acute coronary occlusion [2]. This article summarizes our current understanding of the pathophysiology of these acute coronary syndromes and briefly discusses new approaches currently being researched in an attempt to define and ultimately reduce their incidence.
AMI is usually defined as myocardial necrosis in the setting of clinical evidence consistent with its diagnosis. This clinical evidence includes the patient's symptoms, acute electrocardiograph (ECG) findings, or other evidence indicating a new wall motion abnormality in a segment of the myocardium [3]. Myocardial infarction is caused by an acute imbalance in the ratio of myocardial blood supply to myocardial oxygen demand in the heart. In the case of an acute coronary thrombosis, there is an acute drop in blood flow, leading to myocardial necrosis in the myocardial segment supplied by the coronary artery in question. Sudden cardiac death describes the unexpected natural death from a cardiac cause within a short time period, generally not more than 1 hour from the onset of symptoms, in a person without any prior condition that would appear fatal [4]. SCD is usually secondary to a fatal arrhythmia such as ventricular fibrillation, which is a direct result of the coronary thrombosis lowering the threshold for the onset of this arrhythmia.
Coronary pathophysiology
The underlying pathophysiologic mechanisms for these syndromes begin with the process of atherosclerosis, which develops and progresses for decades prior to the acute event. Atherosclerosis can be described as a low-grade inflammatory state of the intima (inner lining) of medium-sized arteries that is accelerated by the well-known risk factors such as high blood pressure, high cholesterol, smoking, diabetes, and genetics. In the case of coronary atherosclerosis, this slow progression leads to the gradual thickening of the inner layer of the coronary arteries, which may over time narrow the lumen of the artery to various degrees. Atherosclerosis leading to the acute syndromes of AMI and SCD has a predilection for the proximal segments of the major coronary arteries often at arterial bifurcation points that alter flow in the artery [5]. This slow atherosclerotic progression may be interrupted by one or more cycles of rapid progression related to one of two processes: either asymptomatic plaque disruption with formation of a non-occlusive intraluminal thrombus or plaque hemorrhage (Figure 1).
Figure 1. Pathophysiological progression of atherosclerosis.
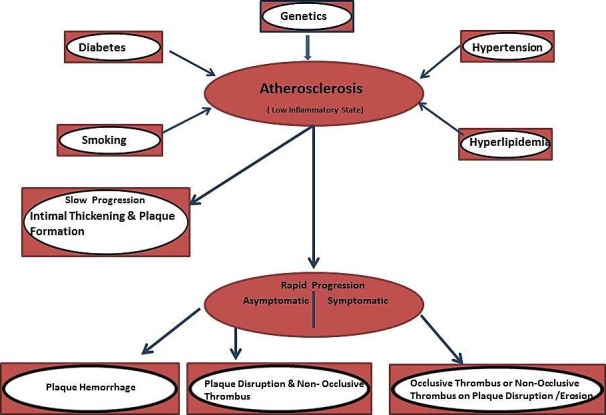
Diagram showing risk factors and progression of atherosclerosis.
Asymptomatic plaque disruption
An atherosclerotic plaque is composed of inflammatory cells, cellular debris, smooth muscle cells (SMCs), and varied amounts of cholesterol and cholesterol ester, some of which is in the form of cholesterol crystals. This lipid core forms in some plaques under a fibrous cap composed of collagen, SMCs, and elastin. The cap is covered on its luminal side by a single layer of endothelial cells as is the inner layer of all arteries within the body. An abundance of inflammatory cells derived foam cells originating from circulating monocytes migrate into the arterial wall and may weaken and thin out the fibrous cap. These plaques are termed thin-capped fibroatheromas (or TCFAs for short) (Figure 2). These processes can ultimately result in a tear in the cap, exposing the thrombogenic lipid core under the cap to the flowing blood, leading to the formation of an intraluminal coronary thrombus. Depending on several factors, including the plaque composition, plaque volume and the degree of luminal narrowing, size of the cap tear, and the thrombotic milieu (a complicated interaction of different forces that ultimately determine how prothrombotic the blood is), the thrombus that forms may either lyse spontaneously, remain, and subsequently be incorporated into the wall of the artery (further narrowing the lumen) or grow and progress to total or near coronary occlusion and a symptomatic acute coronary event [6]. Based on pathologic evaluation of patients with SCD, there are often several bouts of asymptomatic coronary thrombus that have been incorporated into the arterial wall prior to the final fatal event [7].
Figure 2. Thin-capped fibroatheroma.

Diagram shows an artery and the formation of an asymptomatic atherosclerotic plaque.
Plaque erosion is another cause for intraluminal coronary thrombus formation. In this condition, the thrombus forms on a defect in the endothelial layer covering a plaque. These plaques may or may not be inflamed, and the cap is not usually thin. Here, the prothrombotic milieu is felt to be very important in the process, and plaque erosions are common in smokers and in women less than 50 years of age [8]. At present, it is not known how frequently this process occurs asymptomatically. Most of the data on plaque erosion are derived from patients with a symptomatic acute coronary event.
Plaque hemorrhage
As the intima of an artery thickens, it must be nourished by an adequate blood supply. This blood supply or vasa vasorum usually grows from the adventitia or outer layer of the artery into the media and intima supplying needed nutrients. These blood vessels are thin-walled, and their endothelial integrity is not always completely intact. Rupture of these vessels into the intima may acutely enlarge the size of the plaque by the deposition of blood. Furthermore, the red cell membrane is lipid-rich, and hemorrhage can also increase the lipid and inflammatory cell content within the plaque [9].
Thus, slow progression intermixed with cycles of rapid progression leads to plaque growth. However, as the plaque grows, the lumen of the artery does not necessarily narrow. The arterial wall remodels, and the lumen does not begin to narrow until the plaque volume approaches 40% [10]. This phenomenon, called Glagovian or positive remodeling, is in part responsible for the angiographic finding that plaques ultimately responsible for acute coronary events are often non-obstructive (<50% diameter stenosis) in the weeks to month prior to the event [11]. The angiogram visualizes the inside of the artery but cannot evaluate the arterial wall. Whereas the plaque ultimately responsible for the acute event (usually termed vulnerable or high-risk plaques) may be large and bulky, the lumen may look normal or only mildly narrowed on the angiogram because of this remodeling process.
Symptomatic coronary occlusion
Symptomatic coronary occlusion is caused in most cases by intraluminal coronary thrombus formation. Based on post-mortem pathologic evaluation of patients with a fatal coronary thrombosis, plaque rupture is present in about 66% to 75% of cases [12]. Angiograms performed within 4 hours of the onset of AMI symptoms in patients presenting with ST segment elevation on their ECG have shown total occlusion of the coronary artery in 84% of cases, and the remaining cases demonstrated a near total occlusion but with some flow to the distal vessel [13]. The thrombus that forms in many cases is older than the clinical presentation of the AMI. When thrombus was extracted from the responsible coronary at the acute presentation during percutaneous coronary intervention (<12 hours after the onset of symptoms), in about 50% of cases the thrombus that was removed was partially organized, indicating that it formed prior to the onset of symptoms [14].
The underlying pathophysiology of AMI appears to be slightly different when analyzed in the living patient and also depends on the type of AMI. From a clinical standpoint, one divides AMI based on the ECG into ST elevation myocardial infarction (STEMI) and non-ST elevation myocardial infarction (NSTEMI). STEMI in general nearly always presents with total coronary occlusion (see above), and the degree of myocardial necrosis (or the myocardium at risk of necrosis) is larger than in NSTEMI. NSTEMI has a lower incidence of total coronary occlusion, but if the artery lumen is not totally obstructed, it usually demonstrates a severe blockage (>70% diameter stenosis) in one or more arteries most often with an intraluminal coronary thrombus. During evaluations by intracoronary devices at the time of acute presentation to the catheterization laboratory for coronary intervention, differences have been reported between STEMI and NSTEMI in pathophysiology. When ocular coherence tomography was used to evaluate intraluminal pathology, plaque rupture was seen in 72% of STEMI and 32% of NSTEMI and plaque erosion in 28% of STEMI and 48% of NSTEMI. In the remaining NSTEMI cases, a calcified nodule was thought to be the cause [15]. Calcified nodules are found infrequently (<10%) as the underlying mechanism in fatal coronary thrombus at post-mortem examination. Another intraluminal catheter device that can be used during the acute presentation of AMI that detects the presence of lipid-rich plaque has also been studied in patients presenting with STEMI. In 20 patients with an acute coronary occlusion, this catheter found intense yellow plaques (a signal for plaque lipid) in 19 of 20 cases [16].
In search of the vulnerable plaque or patient
If the thrombosed plaque is the immediate cause of most acute coronary events, can one find that plaque that is likely to progress in the future and cause the event? This is the so-called vulnerable or high-risk plaque. As most are non-obstructive and asymptomatic in the weeks prior to the event, relying on symptomatology alone is not useful. However, there is a difference of opinion in the literature as to the best and most cost-effective methods to prevent future acute coronary events. Should one attempt to locate the responsible plaque or concentrate on the high-risk or vulnerable patient likely to develop the acute clinical event?
For vulnerable plaque detection, one would need to find a device, either invasive or (preferably) non-invasive, that could detect the vulnerable plaque. This would necessitate knowing the natural history of such plaques and being able to show that intervening on a presumed vulnerable plaque by some approach such as a stent was safer and more cost-effective than the best medical therapy alone [17]. At present, this is unknown. We still do not know the natural history of a presumed vulnerable plaque, and a plaque that looks vulnerable on an initial evaluation may change at some later point and appear stabilized. However, ongoing studies are trying to address this question. One concern with an invasive methodology is the fact that all of the detectors are concentrating on finding the responsible TCFA but do not consider the plaque erosion which is not an infrequent cause of AMI or SCD. Furthermore, if the detector is invasive, what about individuals in whom the first presentation of symptomatic coronary disease is AMI or SCD? In the Framingham study, 53% of men and 36% of women presented with either AMI or SCD [18]. Non-invasive detectors of a vulnerable plaque are, in our estimation, inadequate and will require additional prospective evaluation.
If vulnerable plaque detection and therapy are challenging, should we concentrate our efforts on the high-risk or vulnerable patient? The answer is probably yes, but similar to the above, this can be difficult, particularly in primary prevention. While patients with known coronary artery disease (CAD) or a CAD equivalent such as diabetes or peripheral artery disease should be treated with guideline-directed medical therapies, how do we identify most of the patients who will ultimately develop AMI or SCD. Traditional risk factor scores are inadequate because, while those at highest risk are more likely to develop adverse events, most adverse events on follow-up do not occur in these patients but in the middle- or lower-risk populations, which are much larger in numbers than the high-risk [19,20]. Various studies assessing different methods of risk evaluation in primary prevention, such as the presence and extent of coronary calcium by computed tomography scanning, intimal-medial thickness of the carotid artery, and iliofemoral atherosclerosis by ultrasound, are attempting to identify that subgroup who might require intensive medical management to reduce subsequent risk. These are ongoing, and results are not yet available [21,22].
In conclusion, our understanding of these acute coronary syndromes has matured over the last 30 years as outlined above. Nevertheless, our methods for detecting and identifying most patients or plaques likely to progress to these syndromes in the future are still inadequate and require further study. It is exciting to speculate, given our past successes, what the next 30 years will reveal.
Abbreviations
- AMI
acute myocardial infarction
- CAD
coronary artery disease
- ECG
electrocardiograph
- NSTEMI
non-ST elevation myocardial infarction
- SCD
sudden cardiac death
- SMC
smooth muscle cell
- STEMI
ST elevation myocardial infarction
- TCFA
thin-capped fibroatheroma
Disclosures
The authors declare that they have no disclosures.
The electronic version of this article is the complete one and can be found at: http://f1000.com/prime/reports/m/7/8
References
- 1.American College of Emergency Physicians; Society for Cardiovascular Angiography and Interventions. O'Gara PT, Kushner FG, Ascheim DD, Casey DE, Jr, Chung MK, de Lemos JA, Ettinger SM, Fang JC, Fesmire FM, Franklin BA, Granger CB, Krumholz HM, Linderbaum JA, Morrow DA, Newby LK, Ornato JP, Ou N, Radford MJ, Tamis-Holland JE, Tommaso CL, Tracy CM, Woo YJ, Zhao DX, Anderson JL, Jacobs AK, Halperin JL, Albert NM, Brindis RG, et al. ACCF/AHA guideline for the management of ST-elevation myocardial infarction: a report of the American College of Cardiology foundation/American Heart Association task force on practice guidelines. J Am Coll Cardiol. 2013;61:e78–140. doi: 10.1016/j.jacc.2012.11.019. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718180330
- 2.Davies M, Thomas AC. Plaque fissuring: the cause of acute myocardial infarction, sudden ischemic death, and crescendo angina. Br Heart J. 1985;53:363–73. doi: 10.1136/hrt.53.4.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thygesen K, Alpert JS, White HD, Joint ESC/ACCF/AHA/WHF Task Force for the Redefinition of Myocardial Infarction. Jaffe AS, Apple FS, Galvani M, Katus HA, Newby LK, Ravkilde J, Chaitman B, Clemmensen PM, Dellborg M, Hod H, Porela P, Underwood R, Bax JJ, Beller GA, Bonow R, Van der Wall EE, Bassand JP, Wijns W, Ferguson TB, Steg PG, Uretsky BF, Williams DO, Armstrong PW, Antman EM, Fox KA, Hamm CW, et al. Universal definition of myocardial infarction. Circulation. 2007;116:2634–53. doi: 10.1161/CIRCULATIONAHA.107.187397. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/723705461
- 4.Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98:2234–51. doi: 10.1161/01.CIR.98.21.2334. [DOI] [PubMed] [Google Scholar]
- 5.Wang JC, Lise S, Normand T, Mauri L, Kuntz RE. Coronary artery spatial distribution of acute myocardial infarction occlusions. Circulation. 2004;110:278–84. doi: 10.1161/01.CIR.0000135468.67850.F4. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/723708689
- 6.Srikanth S, Ambrose JA. Pathophysiology of coronary thrombus formation and adverse consequences of thrombus during PCI. Current Card Rev. 2012;8:168–76. doi: 10.2174/157340312803217247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burke AP, Kolodgie FD, Farb A, Weber DK, Malcom GT, Smialek J, Virmani R. Healed plaque ruptures and sudden coronary death. Circulation. 2001;103:934–40. doi: 10.1161/01.CIR.103.7.934. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/723712598
- 8.Farb A, Burke AP, Tang AL, Liang TY, Mannan P, Smialek J, Virmani R. Coronary plaque erosion without rupture into a lipid core-a frequent cause of coronary thrombosis in sudden coronary death. Circulation. 1996;93:1354–63. doi: 10.1161/01.CIR.93.7.1354. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/723717414
- 9.Kolodgie FD, Gold HK, Burke AP, Fowler DR, Kruth HS, Weber DK, Farb A, Guerrero LJ, Hayase M, Kutys R, Narula J, Finn AV, Virmani R. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–25. doi: 10.1056/NEJMoa035655. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718720485
- 10.Glagov S, Weisenberg E, Zarins C, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316:1371–5. doi: 10.1056/NEJM198705283162204. [DOI] [PubMed] [Google Scholar]
- 11.Ambrose JA, Tannenbaum MA, Alexopoulos D, Hjemdahl-Monsen CE, Leavy J, Weiss M, Borrico S, Gorlin R, Fuster V. Angiographic progression of coronary artery disease and the development of myocardial infarction. J Am Coll Cardiol. 1988;12:56–62. doi: 10.1016/0735-1097(88)90356-7. [DOI] [PubMed] [Google Scholar]
- 12.Falk E, Nakano M, Bentzon JF, Finn AV, Virmani R. Update on acute coronary syndromes: the pathologists’ view. Eur Heart J. 2013;34:719–28. doi: 10.1093/eurheartj/ehs411. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/721226778
- 13.DeWood MA, Spores J, Notske R, Mouser LT, Burroughs R, Golden MS, Lang H. Prevalence of total coronary occlusion during the early hours of transmural myocardial infarction. N Engl J Med. 1980;303:897–902. doi: 10.1056/NEJM198010163031601. [DOI] [PubMed] [Google Scholar]
- 14.Rittersma SZH, Van der Wal AC, Koch KT, Piek JJ, Henriques JP, Mulder KJ, Ploegmakers JP, Meesterman M, de Winter RJ. Instability frequently occurs days or weeks before occlusive coronary thrombosis a pathological thrombectomy study in primary percutaneous coronary intervention. Circulation. 2005;111:1160–5. doi: 10.1161/01.CIR.0000157141.00778.AC. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/723707954
- 15.Jia H, Abtahian F, Aguirre AD, Lee S, Chia S, Lowe H, Kato K, Yonetsu T, Vergallo R, Hu S, Tian J, Lee H, Park SJ, Jang YS, Raffel OC, Mizuno K, Uemura S, Itoh T, Kakuta T, Choi SY, Dauerman HL, Prasad A, Toma C, McNulty I, Zhang S, Yu B, Fuster V, Narula J, Virmani R, Jang IK. In vivo diagnosis of plaque erosion and calcified nodule in patients with acute coronary syndrome by intravascular optical coherence tomography. J Am Coll Cardiol. 2013;62:1748–58. doi: 10.1016/j.jacc.2013.05.071. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718025103
- 16.Madder RD, Goldstein JA, Madden SP, Puri R, Wolski K, Hendricks M, Sum ST, Kini A, Sharma S, Rizik D, Brilakis ES, Shunk KA, Petersen J, Weisz G, Virmani R, Nicholls SJ, Maehara A, Mintz GS, Stone GW, Muller JE. Detection by near-infrared spectroscopy of large lipid core plaques at culprit sites in patients with acute ST-segment elevation myocardial infarction. J Am Coll Cardiol Intv. 2013;6:838–46. doi: 10.1016/j.jcin.2013.04.012. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/725282322
- 17.Ambrose JA. In search of the “Vulnerable Plaque”: can it be localized and will focal regional therapy ever be an option for cardiac prevention? J Am Coll Cardiol. 2008;51:1539–42. doi: 10.1016/j.jacc.2007.12.041. [DOI] [PubMed] [Google Scholar]
- 18.Lerner DJ, Kannel WB. Patterns of coronary heart disease morbidity and mortality in the sexes: a 26 year follow-up of the Framingham population. Am Heart J. 1986;111:383–90. doi: 10.1016/0002-8703(86)90155-9. [DOI] [PubMed] [Google Scholar]
- 19.Wilson PWF, Pencina M, Jacques P, Selhub J, D'Agostino R, Sr, O'Donnell CJ. C-Reactive protein and reclassification of cardiovascular risk in the Framingham heart study. Circ Cardiovasc Qual Outcomes. 2008;1:92–7. doi: 10.1161/CIRCOUTCOMES.108.831198. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718649808
- 20.Akosah KO, Schaper A, Cogbill C. Preventing myocardial infarction in the young adult in the first place. How do the National cholesterol education panel III guidelines perform? J Am Coll Cardiol. 2003;41:1475–9. doi: 10.1016/S0735-1097(03)00187-6. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/725282323
- 21.Falk E, Sillesen H, Muntendam P, Fuster V. The high-risk plaque initiative: primary prevention of atherotrombotic events in the asymptomatic population. Curr Atheroscler Rep. 2011;13:359–66. doi: 10.1007/s11883-011-0193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ortiz AF, Borreguero LJJ, Peñalvo JL, Ordovás JM, Mocoroa A, Fernández-Friera L, Laclaustra M, García L, Molina J, Mendiguren JM, López-Melgar B, de Vega VM, Alonso-Farto JC, Guallar E, Sillesen H, Rudd JH, Fayad ZA, Ibañez B, Sanz G, Fuster V. The progression and early detection of subclinical atherosclerosis (PESA) study: rationale and design. Am Heart J. 2013;166:990–8. doi: 10.1016/j.ahj.2013.08.024. [DOI] [PubMed] [Google Scholar]