

Significance
Cattle were independently domesticated from the aurochs, a wild bovine species, in the vicinity of the current countries of Turkey and Pakistan ∼10,000 y ago. Cattle have since spread with humans across the world, including to regions where these two distinct lineages have hybridized. Using genomic tools, we investigated the ancestry of cattle from across the world. We determined that the descendants of the cattle brought to the New World by the Spanish in the late 1400s show ancestry from multiple domesticated lineages. This pattern resulted from pre-Columbian introgression of genes from African cattle into southern Europe.
Keywords: biogeography, bovine, evolution, genome, introgression
Abstract
Previous archeological and genetic research has shown that modern cattle breeds are descended from multiple independent domestication events of the wild aurochs (Bos primigenius) ∼10,000 y ago. Two primary areas of domestication in the Middle East/Europe and the Indian subcontinent resulted in taurine and indicine lines of cattle, respectively. American descendants of cattle brought by European explorers to the New World beginning in 1493 generally have been considered to belong to the taurine lineage. Our analyses of 47,506 single nucleotide polymorphisms show that these New World cattle breeds, as well as many related breeds of cattle in southern Europe, actually exhibit ancestry from both the taurine and indicine lineages. In this study, we show that, although European cattle are largely descended from the taurine lineage, gene flow from African cattle (partially of indicine origin) contributed substantial genomic components to both southern European cattle breeds and their New World descendants. New World cattle breeds, such as Texas Longhorns, provide an opportunity to study global population structure and domestication in cattle. Following their introduction into the Americas in the late 1400s, semiferal herds of cattle underwent between 80 and 200 generations of predominantly natural selection, as opposed to the human-mediated artificial selection of Old World breeding programs. Our analyses of global cattle breed population history show that the hybrid ancestry of New World breeds contributed genetic variation that likely facilitated the adaptation of these breeds to a novel environment.
The development of genomic tools has given biologists the ability to analyze variation among DNA sequences to reconstruct population history on a fine scale. Given the close interaction of humans with domesticated species and the economic importance of domesticated organisms, it is not surprising that humans have developed many of these species as model organisms. Over the last few years, genomic data have been used to reconstruct the domestication history of many of these species, including dogs (1, 2), horses (3), sheep (4), and cattle (5, 6). The global economic importance of cattle, in combination with the anthropological interest in the shared history of cattle and humans over the last 10,000 y, makes cattle an ideal target for spatial genetic research. The first assembly of the cattle genome sequence was published in 2009 (7, 8). This achievement enables biologists to use genetic variation across breeds and the linkage relationships between those markers to trace the global history of cattle domestication and breed development.
Despite the history of artificial selection in cattle by humans, we report that genomic data can be used to reconstruct broad aspects not only of breed structure but also of the global spatial history of domesticated cattle. Similarly to the strong correlation of genetic variation and geography in European human populations (9), we also find geographic patterning of genetic variation in cattle. Reconstructing the population history of domesticated species is particularly interesting because historical information can be used to realistically constrain parameter estimates in the modeling process. In addition, although the within- versus among-breed partitioning of genetic variation varies widely across different domesticated species (5, 10), most established breeds of cattle can be distinguished using genetic markers (11). Thus, the population history—including movement, population subdivision, hybridization, and introgression—of breeds of domesticated species can be tracked using genetic tools.
Domesticated cattle were introduced to the Caribbean in 1493 by Christopher Columbus, and between 1493 and 1512, Spanish colonists brought additional cattle in subsequent expeditions (12). Spanish colonists rapidly transported these cattle throughout southern North America and northern South America. In the intervening 520 y, they have adapted to the novel conditions in the New World. The descendants of these cattle are known for high feed- and drought-stress tolerance in comparison with other European-derived cattle breeds (13, 14). Genetic variation found within these breeds may be especially valuable in the future adaptation of cattle breeds to climate change. Using genomic tools, we can reconstruct the global population structure of domesticated cattle and determine how different lineages contributed to this group’s evolution.
Domesticated cattle consist of two major lineages that are derived from independent domestications of the same progenitor species, the aurochs (Bos primigenius). The aurochs was a large wild bovine species found throughout Europe and Asia, as well as in North Africa; it has been extinct since 1627 (15). These two primary groups of domesticated cattle are variously treated by different authors as subspecies (Bos taurus taurus and Bos taurus indicus) or as full species (Bos taurus and Bos indicus). For simplicity, we refer here to these two groups as taurine and indicine cattle, respectively. The most obvious phenotypic differences between these groups are the noticeable hump at the withers (i.e., the shoulders of a four-legged mammal) and the floppy rather than upright ears of indicine cattle (16).
The taurine lineage was probably first domesticated in the Middle East, with some later contributions from European aurochsen; the indicine lineage was domesticated on the Indian subcontinent (17). Although archaeological evidence suggests these domestication events likely occurred only 7,000–10,000 y ago (14, 16, 18, 19), there was already preexisting spatial genetic structure in the aurochs population at that time. As a result, the taurine and indicine groups are thought to share a most-recent common ancestor ≥200,000 y ago (20). However, aurochsen and domesticated cattle coexisted in Europe until 1627, and ancient DNA sequencing of aurochs fossils suggests that some large divergences within European domesticated cattle mtDNA may be driven by the repeated incorporation of wild aurochsen into domesticated herds (21, 22). European cattle breeds are largely taurine in origin, whereas cattle from the Indian subcontinent are indicine. Generally, indicine cattle are more feed-stress and water-stress tolerant and are more tropically adapted compared with taurine breeds (23). European taurine cattle have been subjected to more intensive selection for milk and meat production, as well as docility and ease of handling. Taurine and indicine cattle have both contributed genetically to cattle herds in much of Africa (10, 17, 24–26), and microsatellite analyses show a cline of decreasing indicine heritage from east to west and from north to south across the continent (17). Some researchers have suggested that African taurine cattle are derived from a third independent domestication from North African aurochsen (16, 26, 27), although there is also archeological and biological support for postdomestication population structuring within North African herds (18). The major mitochondrial haplogroups within taurine cattle distinguish European from African cattle but show patterns of gene flow north across the Mediterranean, particularly at the Strait of Gibraltar and from Tunisia into Sicily (24, 28). Wild aurochsen in southern Europe and northern Africa, which likely crossed with the domesticated cattle there, may have carried indicine-like haplotypes, but aurochsen mtDNA sampled from Europe to date groups with extant taurine lineages (22).
The first cattle in the Americas were brought to the Caribbean island of Hispaniola, from the Canary Islands, by Christopher Columbus on his second voyage across the Atlantic in 1493, and Spanish colonists continued to import cattle until ∼1512 (13). The descendants of these cattle are the main focus of this paper. The cattle from the Canary Islands were descended from animals of Portuguese and Spanish origin, introduced 20 y earlier by early Spanish explorers (13). Therefore, these cattle likely shared some ancestry with Northern African breeds of cattle and thus may have included an indicine genetic component, via earlier gene flow from Africa to the Iberian Peninsula.
The imported cattle reproduced rapidly in the Caribbean, and by 1512, importation of cattle by ship was no longer necessary (13). Caribbean cattle were introduced into Mexico in 1521 and had been moved north into what is now Texas and south into Colombia and Venezuela within a few decades (13). The Spanish settlers relied on these cattle for meat, but largely allowed them free range in the unfenced wilderness. Artificial selection was occasionally imposed by the choice of which individuals to castrate for steers and which to leave as bulls, except in completely feral herds. Although population sizes plummeted in the late 1800s and herds became more highly managed (13, 29), natural selection had driven the evolution of this group for 400 y (12), or between 80 and 200 generations (30). Although precise generation time of feral populations of cattle is unknown, Texas Longhorns in captivity today reproduce by age 2.
The mostly feral Spanish cattle were the ancestors of the present day New World breeds including Corriente cattle from Mexico, Texas Longhorns from northern Mexico and the southwestern United States, and Romosinuano cattle from Colombia (12). This long period of natural selection left these groups better adapted to these landscapes than breeds of more recent European origin. Texas Longhorns are known to be immune to a tick-borne disease known as “Texas fever” or “Cattle tick fever,” caused by the protozoan Babesia bigemina (31). This pathogen’s vector genus Boophilus is known to have been imported with cattle into the New World (32). Texas Longhorns have also been described to have far greater drought resistance in comparison with more recently imported European breeds (29).
Research on the genetic diversity that was captured by Spanish colonists in the cattle they chose to bring to the New World has been limited. Some African mtDNA haplotypes and microsatellite alleles are also found in Creole (Caribbean) and Brazilian cattle (33). Although some references suggest that cattle may have been brought directly from West Africa to the Caribbean and South America as part of the slave trade, there is no direct historical evidence for this hypothesis (12).
Genomic studies have been conducted on cattle breed population structure (5, 6), but the Iberian lineage of New World cattle has not been investigated in depth. In a phylogenetic analysis on a subset of the single nucleotide polymorphism (SNP) dataset used here, Decker et al. (6) found New World cattle to be the sister group of all other European taurine cattle when heterozygous genotypes were treated as ambiguous characters. However, when genotypes were coded as allele counts (0 for AA, 1 for AB, 2 for BB), the New World cattle were placed within the European clade.
For several hundred years, the only cattle present in North America were those introduced by the Spanish, but indicine cattle were introduced to North America via Jamaica by the 1860s (34). In the mid-1900s, indicine cattle were imported into Brazil, and now there are “naturalized” Brazilian indicine (Nelore) and indicine/taurine hybrid (Canchim) breeds. In some samples of Spanish-derived breeds from South America, mtDNA haplogroups and a Y chromosome microsatellite marker suggest indicine introgression in New World cattle (35, 36). In particular, recent male-mediated introgression of indicine alleles into taurine breeds appears common in Brazil (37).
In this study, we sampled individuals and markers both within New World cattle and from across the globe to study the hybrid history of New World cattle. By analyzing nuclear SNPs scored in cattle from distinct evolutionary lineages, we were able to estimate introgression on a genomic scale. Previous work on New World cattle relied on mtDNA and Y chromosome markers (35). These sequences each reflect the history of a single locus and thus do not have the power to track complex histories of introgression and admixture of genomes. The 47,506 nuclear loci we examined can reflect independent coalescent histories due to recombination and assortment, so they are able to provide much finer resolution of population history than mitochondrial DNA or other single locus markers (38).
Results
Our samples of New World cattle included Texas Longhorn cattle (n = 114), Mexican Corriente cattle (n = 5), and Colombian Romosinuano cattle (n = 8). To place these individuals in a global phylogeographic context, we also included previously published data from individuals of 55 other breeds (n = 1,332; Table 1) (6). These cattle were genotyped for nuclear SNP loci across all 29 autosomal chromosomes using the Illumina BovineSNP50 BeadChip, the Illumina 3K chip, or 6K chip. We analyzed two datasets: one (termed the 1.8k dataset) included 1,814 SNP loci present on all three chips, and the other (termed the 50k dataset) included 47,506 SNP loci from the Bovine SNP50 chip. The 1.8k dataset included more extensive sampling of Texas Longhorn cattle (n = 114) compared with the 50k dataset (n = 40), but a less thorough sampling of the genome.
Table 1.
Breeds included in the analysis
| Figure legend | Name | Region of origin | Sample size 50k (1.8k) |
| 1 | Shorthorn | Great Britain | 99 |
| 2 | Maine Anjou | Southern Europe | 5 |
| 3 | White Park | Great Britain | 4 |
| 4 | Kerry | Great Britain | 3 |
| 5 | Angus | Great Britain | 90 |
| 6 | Devon | Great Britain | 4 |
| 7 | Hereford | Great Britain | 98 |
| 8 | Simmental | Northern Europe | 77 (78) |
| 9 | Red Angus | Great Britain | 15 |
| 10 | Tarentaise | Southern Europe | 5 |
| 11 | Belgian Blue | Northern Europe | 4 |
| 12 | South Devon | Great Britain | 3 |
| 13 | Murray Gray | Australia (via Great Britain) | 4 |
| 14 | English Longhorn | Great Britain | 3 |
| 15 | Red Poll | Great Britain | 5 |
| 16 | Limousin | Southern Europe | 100 |
| 17 | Dexter | Great Britain | 4 |
| 18 | Finnish Ayrshire | Northern Europe | 10 |
| 19 | Guernsey | Channel Islands | 10 |
| 20 | Welsh Black | Great Britain | 2 |
| 21 | Norwegian Red | Northern Europe | 21 |
| 22 | Gelbvieh | Northern Europe | 8 |
| 23 | Scottish Highland | Great Britain | 8 |
| 24 | Pinzgauer | Northern Europe | 5 |
| 25 | Salers | Southern Europe | 5 |
| 26 | Montbeliard | Southern Europe | 5 |
| 27 | Blonde d'Aquitaine | Southern Europe | 5 |
| 28 | Galloway | Great Britain | 4 |
| 29 | Holstien | Northern Europe | 85 (100) |
| 30 | Sussex | Great Britain | 4 |
| 31 | Charolais | Southern Europe | 53 |
| 32 | Belted Galloway | Great Britain | 4 |
| 33 | Brown Swiss | Northern Europe | 10 |
| 34 | Piedmontese | Southern Europe | 29 |
| 35 | Jersey | Channel Islands | 10 |
| 36 | Romagnola | Southern Europe | 29 |
| 37 | Chianina | Southern Europe | 7 |
| 38 | Marchigiana | Southern Europe | 2 (4) |
| 39 | Texas Longhorn | Southwestern United States | 40 (114) |
| 40 | Texas Longhorn cross | Southwestern United States | 5 |
| 41 | Corriente | Mexico | 5 |
| 42 | Romosinuano | Colombia | 8 |
| 43 | Hanwoo Korean | Asia | 7 |
| 44 | Japanese Black | Asia | 10 |
| 45 | Santa Gertrudis | Indicine-taurine hybrid (United States) | 24 |
| 46 | Beefmaster | Indicine-taurine hybrid (United States) | 24 |
| 47 | Senepol | Africa | 36 (37) |
| 48 | N'Dama | Africa | 59 |
| 49 | Tuli | Africa | 4 (5) |
| 50 | Ankole-Watusi | Africa | 5 |
| 51 | N'DamaXBoran | Africa | 42 (41) |
| 52 | Sheko | Africa | 20 |
| 53 | Boran | Africa | 44 |
| 54 | Nelore | Brazil (via India) | 58 (60) |
| 55 | Brahman | United States (via India) | 98 |
| 56 | Guzerat | Brazil (via India) | 3 |
| 57 | Sahiwal | India/Pakistan | 10 |
| 58 | Gir | India | 25 |
Average heterozygosity within breeds ranged from 15% (SD, 1%) in the indicine breed Gir, to 30% (SD, 1%) in the taurine Belgian Blue cattle (Table S1). The highest heterozygosity was 31% (SD, 1%) in the recent hybrid Beefmaster. Generally, as expected from the ascertainment panel for the SNP chip (39), taurine breeds had higher heterozygosity. Breeds of taurine origin averaged heterozygosity of 27%, whereas breeds of indicine origin averaged heterozygosity of 16%. Across New World cattle, average heterozygosity was 28% (Texas Longhorns: 29%, SD, 2%; Corriente; 27%, SD, 2%; Romosinuano: 27%, SD, 1%).
Principal Component Analyses.
For both the 50k and the 1.8k datasets, the first axis of our principal components analysis (PCA) was associated with the indicine–taurine split (Fig. 1; Fig. S1). This axis accounted for 9% of the variance in genotypes in the 1.8k dataset and 13% in the 50k dataset. The second PC axis was associated with the divergence between European and African taurine cattle and accounted for 2.6% (1.8k dataset) to 3.2% (50k dataset) of the variance in genotypes. The placement of African cattle reflected both the gradient of indicine introgression across the continent along PC1 and the divergence between European and African taurine cattle along PC2. N’Dama cattle exhibited the most distinct African taurine ancestry. The New World cattle exhibited intermediate ancestry along both of these axes, with more indicine-like and African-like ancestry than most other European breeds.
Fig. 1.

Statistical summary of genetic variation in 1,461 cattle individuals genotyped at 1,814 SNP loci. Individuals are grouped by the region from which their breed originated, as described in Table 1. Principal component 1 (PC1) captures the split between indicine and taurine domestications. The position of individuals along this axis can be interpreted as the proportion of admixture between these two groups. PC2 captures the European–African split within taurine cattle.
The full 50k SNP dataset overemphasized genetic diversity in British breeds of cattle (especially Herefords; Fig. S1A). Therefore, we reanalyzed the 50k PCA excluding those individuals (Fig. S1B), which resulted in the same patterns seen for the 1.8k data (Fig. 1).
The first 90 PC axes in the 1.8k dataset and the first 154 axes in the 50k dataset were statistically significant based on the Tracy-Widom test (40).
Model-Based Clustering.
In the STRUCTURE analyses of the 1.8k dataset (Fig. 2), we found strong support for two population subdivisions (K), consistent with the deep division of indicine and taurine lineages. The “Hybrid” section shown in Fig. 2 contains the two cattle breeds derived from recent taurine–indicine crosses: Santa Gertrudis (Brahman/Shorthorn) and Beefmaster (Brahman/Hereford/Shorthorn). The STRUCTURE ancestry estimates of these groups reflect their hybrid origins. At K = 2, all New World cattle were estimated to have some indicine ancestry (Fig. 2). Romosinuano cattle from Colombia (n = 8) averaged 14% (SD, 3%) indicine introgression, Corriente cattle from Mexico (n = 5) exhibited 10% (SD, 3%) indicine introgression, and Texas Longhorns (n = 114) averaged 11% (SD, 6%) indicine introgression. An ANOVA showed no significant differences in the extent of indicine introgression among these three groups (P = 0.16).
Fig. 2.
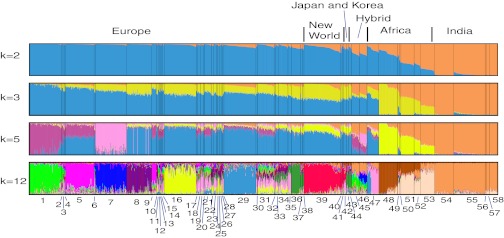
Model-based population assignment for 1,461 individuals based on the 1,814 markers using STRUCTURE (61) and plotted using Distruct (64). Individuals are represented as thin vertical lines, with the proportion of different colors representing their estimated ancestry deriving from different populations. Individuals are grouped by breed as named when sampled; breeds are arranged by regions and are individually labeled by numbers at the bottom. Breed name associated with each number is listed in the “Figure legend” column in Table 1. The best-supported number of ancestral populations was two (K = 2). This split captures the known indicine–taurine split. Hybrid labels refer to Santa Gertrudis (group 45) and Beefmaster (group 46) cattle breeds developed from indicine–taurine crosses within the last 100 y. At K = 3, population groupings were not consistent across runs, but generally followed the division between indicine, European taurine, and African taurine cattle. At higher values of K, individual breed structure predominated, although some breeds (e.g., Jersey, group 35) consistently showed complex ancestry. K = 5 and K = 12 were selected to demonstrate these patterns.
Increasing K beyond two subdivisions resulted in only marginal increases in likelihood scores, which suggested possible model overparameterization. At K = 3, the population subdivisions were roughly consistent with groups of indicine cattle, European taurine cattle, and African cattle (the latter represented by N’Dama cattle; group 48). However, the African subdivision was also present in Mediterranean and New World cattle breeds. At higher values of K, among-breed genetic structure predominated. Levels of indicine introgression varied across individual Texas Longhorns. In agreement with Decker et al. (6), some groups (e.g., Jersey: group 35) consistently showed complex ancestry that was consistent across a range of K values from 3 to 8 (Fig. 2).
Correlation Between Latitude and Genotype.
For breeds originally developed within Europe, we found a significant negative correlation (r = −0.502; P = 0.002) between latitude of country of origin and estimated percent indicine introgression. Percent indicine introgression was estimated from the 1.8k STRUCTURE analyses with K = 2.
Discussion
Simulations have demonstrated that inference of complex historical migration models using PCA is difficult because multiple processes can result in the same patterns (9, 41). Indeed, even under relatively simple scenarios, such as the admixture between two ancestral groups, admixed individuals can be incorrectly assigned to a third group that appears to be geographically intermediate (42). However, when ancestral groups are known, coalescent estimates of admixture between distinct populations are mathematically straightforward. McVean (43) showed that, although PCA is a nonparametric analysis method, coordinates can be predicted from pairwise coalescence times between individuals. This prediction allows a genealogical interpretation to principal component scores. The first principal component can be interpreted as the deepest coalescent event in a tree, and the projection of admixed individuals onto this axis can be used to estimate the proportion of mixture between two parental groups (43). As a test case, we were able to correctly reconstruct the known ancestry of recent taurine–indicine hybrid breeds created for agricultural purposes: Santa Gertrudis (group 45), a Brahman-Shorthorn cross developed in 1918, and Beefmaster (group 46), a cross between Hereford, Shorthorn, and Brahman cattle developed in the 1930s (the “Hybrid” groups shown in Figs. 1 and 2). In addition, we were able to recover the taurine–indicine hybridization cline across Africa along the first principal component (PC1) shown in Fig. 1.
The second principal component (PC2) shown in Fig. 1 separates Eurasian from African cattle, indicating a distinctive genomic component in African breeds. Our samples of African cattle breeds all appear to have admixed taurine–indicine ancestry, based on the intermediate position of African cattle on PC1 (Fig. 1) and the STRUCTURE analyses when K = 2 (Fig. 2). However, the distinctiveness of northern African breeds on PC2 (Fig. 1), as well as in the STRUCTURE analyses when K = 3, indicates additional genomic differentiation in northern African cattle. If African breeds are derived entirely from a mixture of European and Asian cattle, this differentiation must have occurred after the importation of domestic cattle to Africa. Alternatively, this unique African component may be derived from additional domestication events involving north-African aurochsen, as has been suggested previously (16, 26, 27).
Both the principal components analysis (Fig. 1) and the model-based STRUCTURE analyses (Fig. 2; Fig. S1) support a hybrid ancestry for New World cattle, although the patterns of hybridization are distinct from the recently constructed hybrid breeds. New World cattle are largely of taurine descent, but they exhibit an average of 11% indicine ancestry (as estimated from the STRUCTURE analyses of the 1.8k data, with K = 2). In this regard, New World cattle are much like some modern breeds from southern Europe. However, when K = 3 in the STRUCTURE analyses (Fig. 2), much of this “indicine” component in southern European and New World cattle appears to be more specifically associated with cattle from northern Africa. The PCA is also consistent with the hypothesis that New World cattle (as well as modern breeds from southern Europe) are influenced by ancestral gene flow from northern Africa, based on the placement of these breeds at intermediate positions along PC1 and PC2 in Fig. 1.
The pattern of African admixture in southern Europe is consistent with movement of cattle across the Straits of Gibraltar during the Moorish invasion and occupation of the Iberian peninsula in the 8th to 13th centuries CE (6, 18, 44). However, sequencing of Bronze Age cattle mtDNA from Spain suggests that earlier African introgression into Iberia may also have occurred (45). The elevated disease resistance of Texas Longhorn cattle (compared with northern European cattle breeds that have been imported to southwestern North America) may be partially related to the portions of their genomes that stem from this African ancestry. African N’Dama cattle also exhibit some substantial types of disease resistance (46), consistent with this hypothesis.
Using model-based clustering analyses, we found Spanish-derived New World cattle breeds—Texas Longhorns (group 39), Corriente (group 41), and Romosinuano (group 42)—did not differ significantly in levels of indicine introgression (Fig. 3 and Table S1). The Brazilian breeds Nelore (group 54) and Guzerat (group 56) are recently developed breeds from indicine stock, which is reflected in our estimates of their ancestry. All sampled New World cattle that are descended from old Spanish imports (114 Texas Longhorns, 5 Corriente, and 8 Romosinuano) show indicine ancestry (estimated by STRUCTURE, with K = 2). As well, these breeds group together in our PCAs in a position consistent with African introgression. This result suggests that introgression from African cattle occurred before the introduction of these cattle to the New World. This conclusion is supported by the STRUCTURE analysis of the breeds sampled from southern Europe, particularly Italy, which also show indicine and African ancestry. In fact, among the breeds that we sampled, and using the coarse geographical resolution of “country,” we found a significant correlation between latitude in Europe and degree of indicine introgression as estimated from STRUCTURE at K = 2.
Fig. 3.

Geographic structure of breed ancestry as estimated at K = 2 on the 1.8k dataset using STRUCTURE (61). Taurine ancestry is indicated in white and indicine ancestry in black in the pie diagrams. Breed name associated with each number is listed in the “Figure legend” column in Table 1. Note higher levels of indicine introgression in southern Europe, particularly for the Italian breeds Romagnola (group 36), Piedmontese (group 34), Chianina (group 37), and Marchigiana (group 38). The Brazilian breeds Nelore (group 54) and Guzerat (group 56) are recently developed breeds from indicine stock. Pie chart size is scaled to sample size. Breed location is based on the latitude/longitude coordinates from CIA World Factbook (65) of the breed’s country of origin. Silhouettes of cattle are reproduced from ref. 16. This figure was created using the software package GenGIS (66).
The signal of “indicine” introgression in southern Europe may be somewhat misleading, however, depending on the complexity of domestication history in African cattle. In our STRUCTURE analyses at K = 3, the variation captured by the African-like group was not a subset of either of the groups distinguished at K = 2, as would be expected from a strictly bifurcating evolutionary process. The African subdivision at K =3 is at least partly composed of hybrid taurine–indicine genotypes. However, if African cattle are partly derived from a third domestication event involving aurochsen from northern Africa, this deep divergence may be a more important driver of the differentiation between European and African cattle than is indicine introgression. In that case, the indicine component of African and European lineages at K = 2 may reflect African diversity rather than true indicine ancestry.
Additional analyses, including a more thorough sampling of African and Iberian cattle, are needed for a conclusive determination of the number of independent domestication events in cattle. Although our results cannot exclude the possibility of an independent domestication of aurochsen in northern Africa, the relatively low level of variation captured by the second principal component (2–3%) is more consistent with European and African taurine cattle both being derived primarily from a single domestication in the Middle East, with the likely continued but occasional incorporation of genetic material from wild aurochsen in both areas. However, our results do suggest that if there was a third distinct domestication event, it took place in Africa.
Achilli et al. (21) found a novel haplogroup in Italian cattle (Cabannina, not sampled here), for which the timing of divergence was consistent with introgression from European aurochsen. Although continued introgression of aurochs derived genetic material after the original domestication events probably led to greater diversity in European taurine cattle populations, this diversity is not expected to have been indicine-like and therefore is not the likely explanation for the indicine genetic component observed in southern European cattle.
There are at least two alternatives to our interpretation of introgression from Africa into Europe before the introduction of cattle to the New World: (i) relatively recent hybridization with indicine cattle in the New World (within the last 150 y); and (ii) direct importation of cattle from Africa to the Caribbean early in Spanish colonization. Although we cannot completely rule out the possibility of either of these alternatives, neither of these hypotheses explains the signal of shared ancestry between southern Europe and the Americas. Moreover, the first explanation is inconsistent with the clear African-like genomic component in the New World breeds. Finally, the admixture of genomes across chromosomes indicates ancient, rather than recent, introgression. Thus, the simplest explanation is that introgression of genetic material from African cattle occurred before the importation of cattle to the New World by Spanish colonists. The genetic diversity captured by this hybridization likely provided variation for selection when the ancestors of these animals were transported to North America in the late 1400s to early 1500s. However, there is individual variation among Texas Longhorn cattle, with some individuals showing elevated levels of indicine introgression (Fig. 2). This suggests that additional, more recent introgression with indicine cattle may also have occurred in some Texas Longhorn herds.
Our analyses made use of SNP data from across the genome. SNP-chip data have the advantage of being easily replicable, and data reuse across laboratories is straightforward, allowing results to be readily comparable. Furthermore, informative sequence data can inexpensively be generated, allowing investigators to sample many individual cattle. However, it is important to keep in mind the limits of these analyses. As the SNPs selected for the chip were chosen by resequencing individuals on an ascertainment panel, genetic diversity represented in that panel is expected to be overrepresented in future samples (47). In the case of the cattle 50k SNP chip, the ascertainment panel consisted mostly of taurine cattle. The SNPs were selected to be common polymorphisms in these animals (39); therefore, diversity estimates based on these data will overestimate diversity in taurine lineages and underestimate diversity in indicine lineages. Average heterozygosity within groups sampled in this study is consistent with this bias. Because of this bias, we did not attempt to estimate diversity metrics such as FST (48). In addition, the bias toward polymorphisms found in European taurine breeds and for alleles with high minor allele frequencies make these data inappropriate for identifying selective sweeps in New World cattle (39). Although mathematical methods have been developed to correct for ascertainment biases in some cases (49, 50), we did not have the appropriate data regarding the ascertainment process to do so in this case. Nonetheless, McVean (43) showed that, although ascertainment bias has an effect on principal component projections, it does not affect the relative placing of samples. Therefore, ancestry estimation by this method is robust to this source of bias.
Our results are complementary to previous work on the relationships and genetic diversity among cattle breeds (5, 6). Our conclusions match those of the Bovine HapMap Consortium (5) for the breeds that were sampled in both studies.
Our findings of introgression in New World Cattle breeds suggest that European–African admixture (which results in greater apparent divergence) may have driven the apparent sister group relationship between Texas Longhorns and all other European taurine cattle in some analyses presented by Decker et al. (6). Our results also suggest that finding may have resulted from imposing a tree-like structure on populations that arose through complex introgression events.
Although we have only a very sparse sampling of Asian cattle breeds from outside India, our results suggest that these animals are also of hybrid taurine–indicine origin. The possibility of introgression of genetic material from populations or species not sampled in our analysis limits our ability to make inferences about Asian cattle, but they promise to be an interesting area for future research. Although Kawahara-Miki et al. (51) suggested that Japanese cattle are sister to all other domesticated cattle, their omission of an indicine breed in their analyses makes this conclusion difficult to test. In addition, introgression among taurine and indicine lines would produce a similar result in a tree-based analysis.
The recent publication of the first Bos indicus genome sequence (52) will provide an opportunity to identify specific alleles of African or indicine origin that have contributed to the adaptation of New World cattle breeds. Such analyses will be of particular interest given the rapidly changing global climate. New World cattle in general, and Texas Longhorns in particular, are reported to exhibit resilience to drought and harsh climatic conditions (13, 53). Previous work has shown that New World cattle are an important reservoir of genetic diversity (37). As we show here, some of this diversity appears to derive from ancient introgression via African cattle.
Materials and Methods
Sampling.
We examined 1,495 cattle from 58 breeds, including 874 European individuals, 127 individuals from New World breeds, 209 primarily indicine individuals, 260 individuals of African or hybrid origin, and 17 individuals from Japan and Korea (Table S1). A total of 1,420 of these cattle were genotyped for 54,609 single nucleotide loci using the Illumina BovineSNP50 BeadChip (39, 54). We refer to this as the 50k dataset. These data were generated as described by Decker et al. (6). We genotyped an additional 75 Texas Longhorn cattle on one of the Illumina 3K (25 individuals), or 6K (50 individuals) chips. These data were generated commercially at NeoGen/GeneSeek. These individuals were not included in the 50k dataset analysis because the amount of missing data would have greatly exceeded the amount of genotype data. Across the 3k, 6k, and 50k SNP chips are 1,814 shared SNPs, which we refer to as the 1.8k dataset.
Filtering.
We removed SNP loci from our analysis if they were missing from the SNP chip documentation and could not be decoded or identified, if average heterozygosity was >0.5 in 10 or more breeds (which indicated paralogy or repeat regions), or if the call rate was <0.8 in 10 or more breeds (which indicated null alleles or changes in flanking regions preventing DNA hybridization to the array). We also removed markers if they were not found in at least 30% of sampled individuals. We then removed individuals with >10% missing data across the markers on the 29 autosomes from our analyses and subsequently removed markers that were missing in >10% of individuals. A total of 1,369 individuals (Table 1) and 47,506 markers (available on datadyrad.org, provisional DOI: doi:10.5061/dryad.42tr0) were included in the filtered 50k dataset. A total of 1,461 individuals (Table 1) and 1,814 markers were included in the filtered 1.8k dataset. The list of markers in the 50k and 1.8k datasets are included with the data in the Dryad Repository.
To minimize the effects of possible recent hybridization (within the last 150 y), we considered shared genetic signal among Texas Longhorn (United States), Corriente (Mexico), and Romosinuano (Colombia) cattle. We excluded one Texas Longhorn individual from our analyses of New World, because high indicine introgression (∼38%) and large unrecombined chromosomal blocks of indicine ancestry suggested that it was a recent indicine hybrid.
Breed was assigned based on information given by the owner when an individual was sampled. We removed from our analyses two Nelore individuals that do not show any indicine ancestry, strongly suggesting that breed was incorrectly assigned. For all SNPs, we used physical map locations from the University of Maryland assembly of B. taurus, release 3 (7). Geographic locations of breeds were treated at the centroid latitude and longitude of the country from which the breed was known to have originated.
Phasing.
To impute missing data, we required phased haplotype data. Our SNP data were generated as genotype data rather than as haplotype data. Therefore, if an individual was heterozygous at multiple loci, the phase relationship between alleles is not known. We divided our genotype data by chromosome and used a statistical method to phase our genotype data into haplotypes. Genotypes for all individuals in the 50k dataset were phased, and missing data (mean 2%; Table S1) were imputed using fastPHASE (55). We used the defaults of 20 random starts and 25 iterations of the EM algorithm. To avoid biasing haplotype imputation toward preconceived breed structure, we did not use subpopulation identifiers. We allowed fastPHASE to estimate the number of haplotype clusters via a cross-validation procedure described in ref. 55. Pei et al. (56) found fastPHASE to be the most accurate among available genotype imputation software. Imputed genotype data were used only in the PCA.
PCA.
PCA requires complete data, and we therefore performed PCA on imputed, 50k, and 1.8k genotype data. PCA was performed using smartpca in the software package EIGENSOFT (40, 57, 58). The number of significant principal components was calculated using twstats in the eigenstrat package (40). However, Tracy-Widom statistics are estimated based on the assumption of a random sampling of markers, and ascertainment bias in SNPs selected for inclusion on the used SNP chip likely violate this assumption (40, 59).
ANOVA.
ANOVA was performed to test for differences in indicine introgression across New World breeds (Corriente, Romosinuano, and Texas Longhorns), as estimated by the PCA of the 50k and 1.8k datasets and by STRUCTURE. ANOVA was performed in R using aov in the stats package (60).
Model-Based Clustering.
Multilocus model-based clustering, as well as the associated assignment of individuals to populations, was performed using STRUCTURE (61). The SNPs on all 29 autosomes were analyzed using the linkage model based on their UMD3.0 map positions. Recombination rate was treated as uniform. To test for convergence and to aid in parallelization, analyses were repeated five times for each value of K, with a run time of 20,000 iterations and a burn-in of 1,000 iterations. We tested values of K from 2 to 9. In simulations, Evanno et al. (62) found that run lengths >10,000 iterations were not additionally beneficial but that much longer runs still varied in likelihood. We used longer runs as our problem was more complex and tested for convergence across runs after five runs were completed using Structure Harvester (62). STRUCTURE analyses were only conducted on the full unphased 1.8k dataset.
We selected the optimum number of ancestral populations (K) from our STRUCTURE analyses using the method of Evanno et al. (62), implemented in Structure Harvester (63). This method avoids overfitting by selecting the value of K for which there is the largest increase in likelihood from K − 1 to K.
We did not calculate FST values between breeds because the ascertainment in SNP discovery and assay design was strongly biased toward loci common in taurine cattle, which leads to the overestimation of diversity within these breeds.
Supplementary Material
Acknowledgments
We thank Debbie Davis and the Texas Longhorn Cattleman’s Association for research support and genetic samples and Scott Edwards and Robert Wayne for helpful suggestions on the manuscript. E.J.M. was supported by research fellowships awarded by the Graduate Program in Ecology, Evolution, and Behavior at the University of Texas at Austin, Texas EcoLabs, and National Science Foundation BEACON (Bio-Computational Evolution in Action Consortium). This material is based in part on work supported by the National Science Foundation under Cooperative Agreement DBI-0939454. J.F.T., R.D.S., and J.E.D. were supported by National Research Initiative Grants 2008-35205-04687 and 2008-35205-18864 from the US Department of Agriculture (USDA) Cooperative State Research, Education and Extension Service and National Research Initiative Grants 2009-65205-05635 and 2012-67012-19743 from the USDA National Institute of Food and Agriculture.
Footnotes
The authors declare no conflict of interest.
Data deposition: The single nucleotide polymorphism data reported in this paper have been deposited in the Dryad Data Repository, http://dx.doi.org/10.5061/dryad.42tr0.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303367110/-/DCSupplemental.
References
- 1.Larson G, et al. Rethinking dog domestication by integrating genetics, archeology, and biogeography. Proc Natl Acad Sci USA. 2012;109(23):8878–8883. doi: 10.1073/pnas.1203005109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vonholdt BM, et al. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature. 2010;464(7290):898–902. doi: 10.1038/nature08837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Achilli A, et al. Mitochondrial genomes from modern horses reveal the major haplogroups that underwent domestication. Proc Natl Acad Sci USA. 2012;109(7):2449–2454. doi: 10.1073/pnas.1111637109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kijas JW, et al. International Sheep Genomics Consortium Members Genome-wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012;10(2):e1001258. doi: 10.1371/journal.pbio.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gibbs RA, et al. Bovine HapMap Consortium Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science. 2009;324(5926):528–532. doi: 10.1126/science.1167936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Decker JE, et al. Resolving the evolution of extant and extinct ruminants with high-throughput phylogenomics. Proc Natl Acad Sci USA. 2009;106(44):18644–18649. doi: 10.1073/pnas.0904691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimin AV, et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009;10(4):R42. doi: 10.1186/gb-2009-10-4-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elsik CG, et al. Bovine Genome Sequencing and Analysis Consortium The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science. 2009;324(5926):522–528. doi: 10.1126/science.1169588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Novembre J, et al. Genes mirror geography within Europe. Nature. 2008;456(7218):98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freeman AR, et al. Admixture and diversity in West African cattle populations. Mol Ecol. 2004;13(11):3477–3487. doi: 10.1111/j.1365-294X.2004.02311.x. [DOI] [PubMed] [Google Scholar]
- 11.Kuehn LA, et al. Predicting breed composition using breed frequencies of 50,000 markers from the US Meat Animal Research Center 2,000 Bull Project. J Anim Sci. 2011;89(6):1742–1750. doi: 10.2527/jas.2010-3530. [DOI] [PubMed] [Google Scholar]
- 12.Rouse JE. The Criollo: Spanish Cattle in the Americas. Norman, OK: Univ of Oklahoma Press; 1977. [Google Scholar]
- 13.Barragy TJ. Gathering Texas Gold. Cayo del Grullo, TX: Cayo Del Grullo Press; 2003. [Google Scholar]
- 14.Clutton-Brock J. A Natural History of Domesticated Mammals. Cambridge, UK: Cambridge Univ Press; 1999. [Google Scholar]
- 15.Mona S, et al. Population dynamic of the extinct European aurochs: Genetic evidence of a north-south differentiation pattern and no evidence of post-glacial expansion. BMC Evol Biol. 2010;10:83. doi: 10.1186/1471-2148-10-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grigson C. An African origin for African cattle?—Some archaeological evidence. Afr Archaeol Rev. 1991;9:119–144. [Google Scholar]
- 17.MacHugh DE, Shriver MD, Loftus RT, Cunningham P, Bradley DG. Microsatellite DNA variation and the evolution, domestication and phylogeography of taurine and zebu cattle (Bos taurus and Bos indicus) Genetics. 1997;146(3):1071–1086. doi: 10.1093/genetics/146.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loftus RT, MacHugh DE, Bradley DG, Sharp PM, Cunningham P. Evidence for two independent domestications of cattle. Proc Natl Acad Sci USA. 1994;91(7):2757–2761. doi: 10.1073/pnas.91.7.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perkins D., Jr Fauna of Catal Hüyük: Evidence for early cattle domestication in Anatolia. Science. 1969;164(3876):177–179. doi: 10.1126/science.164.3876.177. [DOI] [PubMed] [Google Scholar]
- 20.Hiendleder S, Lewalski H, Janke A. Complete mitochondrial genomes of Bos taurus and Bos indicus provide new insights into intra-species variation, taxonomy and domestication. Cytogenet Genome Res. 2008;120(1–2):150–156. doi: 10.1159/000118756. [DOI] [PubMed] [Google Scholar]
- 21.Achilli A, et al. The multifaceted origin of taurine cattle reflected by the mitochondrial genome. PLoS ONE. 2009;4(6):e5753. doi: 10.1371/journal.pone.0005753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bailey JF, et al. Ancient DNA suggests a recent expansion of European cattle from a diverse wild progenitor species. Proc Biol Sci. 1996;263(1376):1467–1473. doi: 10.1098/rspb.1996.0214. [DOI] [PubMed] [Google Scholar]
- 23.Frisch J, Vercoe J. Food intake, eating rate, weight gains, metabolic rate and efficiency of feed utilization in Bos taurus and Bos indicus crossbred cattle. Anim Prod. 1977;25(3):343–358. [Google Scholar]
- 24.Cymbron T, Loftus RT, Malheiro MI, Bradley DG. Mitochondrial sequence variation suggests an African influence in Portuguese cattle. Proc Biol Sci. 1999;266(1419):597–603. doi: 10.1098/rspb.1999.0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loftus RT, et al. A microsatellite survey of cattle from a centre of origin: The Near East. Mol Ecol. 1999;8(12):2015–2022. doi: 10.1046/j.1365-294x.1999.00805.x. [DOI] [PubMed] [Google Scholar]
- 26.Hanotte O, et al. African pastoralism: Genetic imprints of origins and migrations. Science. 2002;296(5566):336–339. doi: 10.1126/science.1069878. [DOI] [PubMed] [Google Scholar]
- 27.Bradley DG, MacHugh DE, Cunningham P, Loftus RT. Mitochondrial diversity and the origins of African and European cattle. Proc Natl Acad Sci USA. 1996;93(10):5131–5135. doi: 10.1073/pnas.93.10.5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beja-Pereira A, et al. The origin of European cattle: Evidence from modern and ancient DNA. Proc Natl Acad Sci USA. 2006;103(21):8113–8118. doi: 10.1073/pnas.0509210103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobie JF. The Longhorns. Austin, TX: Univ of Texas Press; 1980. [Google Scholar]
- 30.Kantanen J, et al. Temporal changes in genetic variation of north European cattle breeds. Anim Genet. 1999;30(1):16–27. doi: 10.1046/j.1365-2052.1999.00379.x. [DOI] [PubMed] [Google Scholar]
- 31.Figueroa JV, Chieves LP, Johnson GS, Buening GM. Detection of Babesia bigemina-infected carriers by polymerase chain reaction amplification. J Clin Microbiol. 1992;30(10):2576–2582. doi: 10.1128/jcm.30.10.2576-2582.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.George JE, Davey RB, Pound JM. Introduced ticks and tick-borne diseases: The threat and approaches to eradication. Vet Clin North Am Food Anim Pract. 2002;18(3):401–416, vi. doi: 10.1016/s0749-0720(02)00030-0. [DOI] [PubMed] [Google Scholar]
- 33.Magee DA, et al. A partial african ancestry for the creole cattle populations of the Caribbean. J Hered. 2002;93(6):429–432. doi: 10.1093/jhered/93.6.429. [DOI] [PubMed] [Google Scholar]
- 34. Hoyt AM (1982) History of Texas Longhorns. Texas Longhorn J 1982:1–48. Available at http://doublehelixranch.com/History.html.
- 35.Ginja C, et al. Origins and genetic diversity of New World Creole cattle: Inferences from mitochondrial and Y chromosome polymorphisms. Anim Genet. 2010;41(2):128–141. doi: 10.1111/j.1365-2052.2009.01976.x. [DOI] [PubMed] [Google Scholar]
- 36.Mirol PM, Giovambattista G, Lirón JP, Dulout FN. African and European mitochondrial haplotypes in South American Creole cattle. Heredity (Edinb) 2003;91(3):248–254. doi: 10.1038/sj.hdy.6800312. [DOI] [PubMed] [Google Scholar]
- 37.Giovambattista G, et al. Male-mediated introgression of Bos indicus genes into Argentine and Bolivian Creole cattle breeds. Anim Genet. 2000;31(5):302–305. doi: 10.1046/j.1365-2052.2000.00658.x. [DOI] [PubMed] [Google Scholar]
- 38.Edwards S, Bensch S. Looking forwards or looking backwards in avian phylogeography? A comment on Zink and Barrowclough 2008. Mol Ecol. 2009;18(14):2930–2933, discussion 2934–2936. doi: 10.1111/j.1365-294X.2009.04270.x. [DOI] [PubMed] [Google Scholar]
- 39.Matukumalli LK, et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE. 2009;4(4):e5350. doi: 10.1371/journal.pone.0005350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.François O, et al. Principal component analysis under population genetic models of range expansion and admixture. Mol Biol Evol. 2010;27(6):1257–1268. doi: 10.1093/molbev/msq010. [DOI] [PubMed] [Google Scholar]
- 42.Novembre J, Stephens M. Interpreting principal component analyses of spatial population genetic variation. Nat Genet. 2008;40(5):646–649. doi: 10.1038/ng.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McVean G. A genealogical interpretation of principal components analysis. PLoS Genet. 2009;5(10):e1000686. doi: 10.1371/journal.pgen.1000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis SJM. Zooarchaeological evidence for Moslem and Christian improvements of sheep and cattle in Portugal. J Archaeol Sci. 2008;35:991–1010. [Google Scholar]
- 45.Anderung C, et al. Prehistoric contacts over the Straits of Gibraltar indicated by genetic analysis of Iberian Bronze Age cattle. Proc Natl Acad Sci USA. 2005;102(24):8431–8435. doi: 10.1073/pnas.0503396102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murray M, Trail JC, Davis CE, Black SJ. Genetic resistance to African trypanosomiasis. J Infect Dis. 1984;149(3):311–319. doi: 10.1093/infdis/149.3.311. [DOI] [PubMed] [Google Scholar]
- 47.Albrechtsen A, Nielsen FC, Nielsen R. Ascertainment biases in SNP chips affect measures of population divergence. Mol Biol Evol. 2010;27(11):2534–2547. doi: 10.1093/molbev/msq148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nielsen R. Population genetic analysis of ascertained SNP data. Hum Genomics. 2004;1(3):218–224. doi: 10.1186/1479-7364-1-3-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuhner MK, Beerli P, Yamato J, Felsenstein J. Usefulness of single nucleotide polymorphism data for estimating population parameters. Genetics. 2000;156(1):439–447. doi: 10.1093/genetics/156.1.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y, Nielsen R. Estimating population divergence time and phylogeny from single-nucleotide polymorphisms data with outgroup ascertainment bias. Mol Ecol. 2012;21(4):974–986. doi: 10.1111/j.1365-294X.2011.05413.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawahara-Miki R, et al. Whole-genome resequencing shows numerous genes with nonsynonymous SNPs in the Japanese native cattle Kuchinoshima-Ushi. BMC Genomics. 2011;12:103. doi: 10.1186/1471-2164-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canavez FC, et al. Genome sequence and assembly of Bos indicus. J Hered. 2012;103(3):342–348. doi: 10.1093/jhered/esr153. [DOI] [PubMed] [Google Scholar]
- 53.Riely A. The grass-fed cattle-ranching niche in Texas. Geogr Rev. 2011;101(2):261–268. [Google Scholar]
- 54.Van Tassell CP, et al. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat Methods. 2008;5(3):247–252. doi: 10.1038/nmeth.1185. [DOI] [PubMed] [Google Scholar]
- 55.Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data: Applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet. 2006;78(4):629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pei Y-F, Li J, Zhang L, Papasian CJ, Deng H-W. Analyses and comparison of accuracy of different genotype imputation methods. PLoS ONE. 2008;3(10):e3551. doi: 10.1371/journal.pone.0003551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Price AL, et al. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 2009;5(6):e1000519. doi: 10.1371/journal.pgen.1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 59.Tracy CA, Widom H. Level-spacing distributions and the Airy kernel. Commun Math Phys. 1994;159:151–174. [Google Scholar]
- 60.R Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2010. [Google Scholar]
- 61.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol Ecol. 2005;14(8):2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 63.Earl DA, Vonholdt BM. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genet Resour. 2012;4(2):359–361. [Google Scholar]
- 64.Rosenberg NA. DISTRUCT: A program for the graphical display of population structure. Mol Ecol Notes. 2003;4(1):137–138. [Google Scholar]
- 65.Central Intelligence Agency . The World Factbook. Washington: Central Intelligence Agency; 2008. Available at https://www.cia.gov/library/publications/the-world-factbook/. Accessed October 5, 2011. [Google Scholar]
- 66.Parks DH, et al. GenGIS: A geospatial information system for genomic data. Genome Res. 2009;19(10):1896–1904. doi: 10.1101/gr.095612.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.