

Abstract
Molecular biology has become heavily dependent on biological knowledge encoded in expert curated biological databases. As the volume of biological literature increases, biocurators need help in keeping up with the literature; (semi-) automated aids for biocuration would seem to be an ideal application for natural language processing and text mining. However, to date, there have been few documented successes for improving biocuration throughput using text mining. Our initial investigations took place for the workshop on ‘Text Mining for the BioCuration Workflow’ at the third International Biocuration Conference (Berlin, 2009). We interviewed biocurators to obtain workflows from eight biological databases. This initial study revealed high-level commonalities, including (i) selection of documents for curation; (ii) indexing of documents with biologically relevant entities (e.g. genes); and (iii) detailed curation of specific relations (e.g. interactions); however, the detailed workflows also showed many variabilities. Following the workshop, we conducted a survey of biocurators. The survey identified biocurator priorities, including the handling of full text indexed with biological entities and support for the identification and prioritization of documents for curation. It also indicated that two-thirds of the biocuration teams had experimented with text mining and almost half were using text mining at that time. Analysis of our interviews and survey provide a set of requirements for the integration of text mining into the biocuration workflow. These can guide the identification of common needs across curated databases and encourage joint experimentation involving biocurators, text mining developers and the larger biomedical research community.
Introduction
We summarize here our findings stemming from a workshop on ‘Text Mining for the BioCuration Workflow,’ held at the third International Biocuration Conference (Berlin, 2009). The workshop goals were to bring together text mining developers with biological biocurators in order to:
facilitate cross-education, so that biocurators would have a better understanding of the capabilities and limitations of text mining, and the text mining developers would better understand the requirements of the biocuration community and
identify good candidate technologies and insertion points for those technologies in the biocuration workflow.
In preparation for the workshop, the organizers interviewed biocurators from eight expert curated biological databases to develop a more detailed understanding of biocuration workflows. The workshop included introductory presentations by the organizers, contributed talks on experiences inserting text mining tools into the curation workflow, and a well-attended discussion session. Following the workshop, the organizers, in cooperation with Pascale Gaudet and the International Society for Biocuration (http://www.biocurator.org/), undertook a survey of biocurators’ needs and experiences related to text mining in the biocuration workflow.
The article is organized as follows: the Background section introduces the challenges faced by the biocuration community in maintaining a growing number of biological databases; the next two sections describe the biocuration workflow and the text mining approaches that have been applied to address the biocuration issues. The section on Findings describes findings from the pre-workshop survey of biocuration workflows, the workshop discussion and the results from the post-workshop survey of biocurators. The two final sections discuss the findings and outline next steps, including follow-on workshops to address the major findings described in the article.
Background
Biological databases serve to collect and provide access to our expanding knowledge of biology. The number of biological databases increases every year: the 2011 Nucleic Acid Research Database issue (1) reports that there are now over 1300 biological databases, 96 of them new in 2011. In today's age of massive data sets, high-throughput experiments and multi-disciplinary research, we need more efficient ways of accessing and ‘digesting’ biological information into computable form.
There are several possible ways to achieve this. One approach is to require authors to deposit data in a repository—the GenBank model. The advantage is that the expense of adding to the database is spread across all the researchers who contribute to the formation of biological knowledge. The disadvantages are that the quality of the data captured can be variable, and the data are often incomplete.
At the other end of the spectrum is expert biocuration. This approach provides high-quality entries, but is expensive to maintain; examples include the model organism, protein, pathway and interaction databases. Since this approach relies on trained expert biocurators who read and extract ‘curatable’ information from the published literature, curation can become a potential bottleneck, both in terms of speed and cost. Text mining tools have the potential to speed up the curation process if they perform useful tasks with sufficient accuracy and speed. We undertook this study in order to identify ways in which text mining tools could help, and where such tools could be usefully inserted into the curation process.
The biocuration workflow
Literature curation requires a careful examination by domain experts of relevant literature descriptions from the scientific literature, extracting essential information in a formalized way to fill in structured database records. Biocuration workflows have been increasingly used in the bioinformatics domain to enable reproducible analysis of biological data by means of computational tools (2,3). Such workflows are similar to descriptions of methods in experimental research whose purpose is to facilitate reproducibility of the findings and enable interpretation of their significance. In addition, documenting the workflow captures the state of the practice. This would enable newer databases to develop their workflows and guidelines more efficiently, and could, in turn, lead to better documentation and faster training of new biocurators.
Based on the interactions with a number of biocuration groups and a set of interviews conducted prior to the workshop, we identified a ‘canonical’ biocuration workflow, consisting of the following stages (Figure 1).
(A) Triage: finding curation relevant articles.
(B) Bio-entity identification and normalization: detecting mentions of bio-entities of relevance for curation, e.g. genes, proteins or small molecules, linked to unique database identifiers, such as those in UniProt, EntrezGene or ChEBI.
(C) Annotation event detection: identifying and encoding annotatable events, such as descriptions of protein–protein interactions, characterizations of gene products in terms of their cellular location, their molecular function, biological process involvement and phenotypic effect.
(D) Evidential qualifier association: association of experimental evidence supporting the annotation event carried out as a result of biocuration efforts.
(E) Database record completion and check.
Figure 1.

Text mining and the biocuration workflow: main tasks of a canonical annotation workflow, including (A) triage, (B) bio-entity identification and normalization, (C) annotation event detection, (D) evidential qualifier association and (E) database record completion.
In practice, these tasks are often interconnected and interleaved with additional intermediate steps that may change the order of activities; for example, one workflow may require prior annotation of evidence before carrying out the entity normalization. Curation can be ‘entity-based’, where the curation team prioritizes papers for a certain class of entity, e.g. all papers on a particular gene or chemical; or the strategy can be journal-based (all the papers published in the last month in a set of journals) or novelty-based (e.g. not yet curated entities or novel functions) or a combination of such considerations.
The specific tasks in the workflow may be dependent on maturity of the database, on the volume of literature to be curated and on the size of the biocuration staff. In early phases, the database may be the product of a single person, and the workflow may evolve rapidly. For mature databases, the curation workflow may be complex, with extensive documentation, with detailed curation/annotation guidelines to ensure consistency across a team of biocurators. Biological databases also vary in size. For a small database, typically a few biocurators do all the steps; for the larger databases, the biocuration staff may be more specialized to cover specific activities. These issues are explored further in Supplementary Appendix A1, and more details are provided in (4,5).
Text mining for the biocuration workflow
Text mining applications for the biocuration workflow can be divided into broad categories that correspond roughly to the subtasks shown in Figure 1. Task A (triage) relies on information retrieval, sometimes known as text categorization. This step involves binning documents (articles) into ‘curatable’ documents versus those not needing to be curated. This step may also involve a prioritization or ranking of documents, with documents containing information on novel discoveries (genes, proteins interactions) assigned a higher priority.
Once relevant documents have been retrieved, the next step is to determine what things of interest are mentioned in them (Task B: bio-entity identification and normalization). Here, there are two separate but related tasks. The first step is entity-tagging that involves identifying mentions of biological entities of interest in the text. A common example of this is known as gene mention that involves finding every location in the article where a gene is mentioned by name. The second step involves normalization that links the mentions of a biological entity to its unique identifier in the appropriate resource. For example, for gene normalization, gene mentions are linked to their unique gene identifiers in an accepted external resource such as EntrezGene, producing the set of gene identifiers for genes mentioned in the article. These tasks can be generalized to other kinds of bio-entities, such as proteins (linked to UniProt identifiers), organisms, chemicals and small molecules.
Relation extraction supports the ability to identify specific relations among entities in the document (Task C: Annotation event detection). For example, if two proteins are mentioned, are they involved in a protein–protein interaction? Systems that do this are among the most tantalizing products of text mining, but they are also probably the least advanced. Finally, evidence extraction (Task D: Evidential qualifier association) is of critical importance, allowing biologists to link an annotation to the corresponding evidence, as it appears in the article. This task is particularly challenging to evaluate, but is key to biologists’ ability to assess the information provided in biological databases. Challenges include the fact that evidence may be spread throughout the article, and may also be repeated in multiple places, such as in a figure legend and in the associated text. This makes it difficult to evaluate system performance based on whether it has found the right evidence (or ‘good enough’ evidence).
Text mining for the genomics domain has been a topic of research for at least 10 years—see for example refs (6–9). In that period, text mining has been able to achieve success rates in the range of 90% for specific narrowly defined ‘stand-alone’ tasks, such as gene mention identification in running text (10). Related to this research, there have been a number of open challenge evaluations that have allowed multiple groups to compare their results on specific problems, such as prioritization of articles for curation, or extraction of biological entities of interest.
Evaluations have included BioCreative [Critical Assessment for Information Extraction in Biology] (10–12), the TREC Genomics track (13,14) and the BioNLP (natural language processing) for biology tasks associated with the Association for Computational Linguistics, e.g. (15). To date, these evaluations have focused on isolated text mining tasks, following the tradition established in the natural language processing community that has emphasized ‘off-line’ accuracy measures, such as precision, recall and balanced F-measure (the harmonic mean of precision and recall). These measures explicitly avoid having a human in the loop, and are thus useful for iterative and repeatable evaluations. The disadvantage of such evaluations is, however, that there has been little focus on interactive, biocurator-centric tools and limited formal evaluation of tools in terms of whether they can assist biocurators. That said, there is increasing focus on creation of interactive tools for curation; see for example, (16), or papers from the text mining session at the 2008 Pacific Symposium for Biocomputing on ‘Translating Biology: Text Mining Tools that Work’ (17,18).
Table 1 shows a partial list of available text mining tools and their potential contributions to the different stages of biocuration workflow. Of the text mining systems listed in the table, only Textpresso (6) has achieved significant adoption in the production of biological database systems (see discussion in Findings below). For the triage task (A), a variety of text mining tools have the potential to increase the throughput of expert biocurators by helping to identify or prioritize articles for curation (7,8). For tasks B and C (entity linkage, annotation event detection), there is evidence that tools can assist the biocurator in encoding the critical information by linking biological entities (e.g. genes, proteins) to reference databases, such as EntrezGene or UniProt. These tools can also be used to improve completeness of author-deposited information (9–11). The recent BioCreative III evaluation (12) focused on the potential of interactive systems to assist the biocurator in identifying and linking ‘curatable’ biological entities. Task D (Evidential qualifier association) was evaluated in BioCreative II (13), but to date, only a few tools have been able to address this aspect of curation, which generally requires extraction of relations among entities.
Table 1.
Partial list of text mining tools and capabilities in the BioCuration Workflow supporting: Triage, bio-entity identification and normalization, annotation relation and event detection and evidential qualifier association
 |
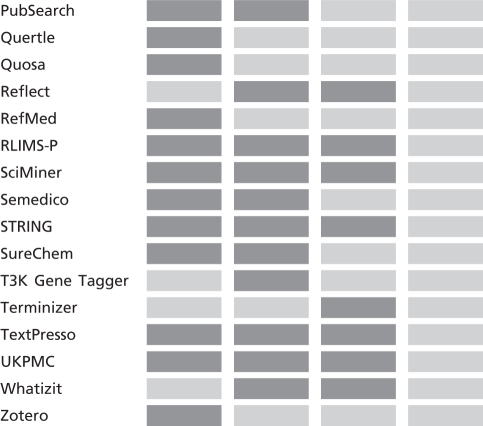 |
A dark cell indicates that the tool is applicable to the task; a light color cell indicates not applicable. Tools are linked to their associated website
In parallel to the development of research tools, there has also been the development of robust, commercial quality tools for use at pharmaceutical (pharma) and biotechnology companies, such as the suites developed by Ariadne (www.ariadnegenomics.com) and Linguamatics (www.linguamatics.com). Adopters of these commercial tools can realize savings because of the scale of their operations, despite significant investment to purchase the tools. However, such commercial grade tools are generally beyond the budget of most publicly maintained biological databases, which are typically funded by grants with limited resources to invest in ‘infrastructure’.
Findings
To identify biocuration requirements, we carried out a detailed analysis of biocuration pipelines in preparation for the workshop. At the workshop, there were presentations from the organizers, but also from a number of groups experimenting with text mining and the curation workflow. The workshop also included a discussion session where biocurators and developers were able to discuss the challenges from the perspectives of text mining and biocuration. Following the workshop, we surveyed biocurators on current annotation processes, priorities and existing bottlenecks.
Pre-workshop analysis of biocuration workflows
A major stumbling block for the application of text mining tools is the need to integrate any new tool into the curation workflow, and to tailor it to produce the kind of output needed by that database. To understand better the needs from the perspective of the biocurators, we undertook a detailed study of the biocuration workflow for eight biological databases listed in *bold in Table 2. Members of the team (G.A.P.C.B., M.K., C.A., K.B.C) interviewed biocurators, and G.A.P.C.B. encoded the workflows in formal modeling language (UML) (see Supplementary Appendix A1).
Table 2.
Biological databases represented in the surveys: biocurators from databases in *bold were interviewed for the initial biocuration workflow study
| Description | |
|---|---|
| Protein–protein interaction | |
| *BioGRID | Physical and genetic interactions |
| *MINT | Physical interactions |
| Model Organism Databases | |
| *SGD | Saccharomyces Genome Database |
| RGD | Rat Genome Database |
| *TAIR | Arabidopsis Genome Database |
| *MGI | Mouse Genome Informatics Datbase |
| Dictybase | Dictyostelium discoideum genome database |
| MaizeGDB | Maize Genome Database |
| WormBase | Database of the biology and genome of C. Elegans |
| FlyBase | Database of Drosophila genetics and molecular biology |
| SoyBase | Resource for soybean researchers |
| Protein | |
| UniProt | Protein Database |
| *PRO | Protein Ontology |
| Pathway and reactions | |
| Reactome | Signaling and metabolic pathway focused on Human |
| *Gallus | Signaling and metabolic pathway focused on chicken |
| SABIO-RK | SABIO-Reaction Kinetics Database Genome |
| Others | |
| JGI | Joint Genome Institute genome portal |
| *Comparative Toxicogenomics Database | Gene–disease–chemical interactions database |
| AgBase | Resource for functional analysis of agricultural plant and animal gene products |
| @NoteWiki | Genome-scale metabolic reconstruction and regulatory network analysis |
| Cardiovascular Gene Ontology | Gene Ontology annotations for the cardiovascular system |
| modENCOD | Model organism ENCyclopedia Of DNA Elements project |
| BioWisdom | Healthcare intelligent system |
Our series of interviews showed that detailed workflow differs from database to database, reflecting differences in requirements, volume of literature to be curated, length of time the databases have been operating and the scale and complexity of the curation activities. For example, some databases require the annotation of additional entities and relations relevant to the experimental setup, such as tissue types and cell lines, as well as patient-related information. Many databases only curate findings that have experimental evidence provided in the article. Access to and processing of textual materials may be a problem, particularly for tables and figures, and for information provided in the article's supplementary material.
Workshop and follow-up
At the workshop, the organizers summarized their findings on biocuration workflows and provided an overview of text mining terms and methods. This was followed by talks addressing practical experiences applying text mining to biocuration focused on two themes: people who had built tools that had the potential to make a contribution to biocuration work, by Lourenço (14) and Wiegers (7); also two groups reported on their successes and failures in applying text mining to biocuration work: Dowell (15) and Veuthey (16). In addition, Chatr-aryamontri reported on an experiment with author curation for the MINT database (17) and Cohen presented Bada and Hunter's talk on annotation (18). The final segment of the workshop was devoted to discussion, including an informal poll to get a biocurator ‘wish list’. A number of biocurators expressed interest in having text mining tools capture other kinds of information, such as phenotype, chemicals or Gene Ontology (GO) terms.
Survey of biocurators
The initial pre-workshop interviews with curators and the lively discussion at the well-attended workshop at the Biocuration Conference motivated the workshop organizers to explore further the integration of text mining into the biocuration workflow. Following the workshop, the organizers put together a survey on current annotation processes and existing bottlenecks, in order to get more detailed insight into biocuration practices, experiences with text mining and priorities for new tools from the biocurator perspective.
The survey covered four areas: (i) information about the curator and curation task; (ii) information about the curation workflow, including article selection, strategy for curating individual abstracts/articles and bio-entities to be captured; (iii) experiences with text mining tools; and (iv) curator requirements or wish list for text mining tools. The survey is discussed in detail in Supplementary Appendix B1 and the responses are provided in Supplementary Appendix C1.
Overall, there were 30 respondents from 23 databases and other resources (Table 2). The key findings from the responses to the survey were as follows:
biocurators are adopters of text mining technology. Over 70% had tried text mining, and almost 50% were using it in some form. The most widely used system was Textpresso, with 7 out of 28 curators using it for some aspect of curation (survey question 8);
the application of greatest interest to curators was document selection and prioritization: 19 out of 27 curators responded that they make or would make heavy (14) or moderate (5) use of text mining for this purpose (survey question 9);
identification of underlying evidence was also of great interest: 19 out of 27 curators would make heavy (9) or moderate (10) use of this (survey question 9); and
aids to link biological entities to underlying biological resources, including ontological resources were also of high interest: curators would make heavy (8) or moderate (10) use of aids to link to resources such as EntrezGene or GO (survey question 9).
The survey also identified a number of interesting issues including the following.
Ability to handle full text was a top priority; 27 out of 29 respondents curated from full text routinely (21) or as needed (6) (survey question 4). The need to handle full text imposed related requirements, including ability to handle multiple file formats (Microsoft Word .doc, Adobe Acrobat .pdf, Excel .xls), as well as access to and persistence of supplementary materials.
Curation from figures and tables was a standard practice (23 and 24 out of 24 respondents, respectively, in response to survey question 5).
Ontologies and standardized terminologies are in widespread use across diverse organisms and tasks. For example, 23 out of 29 respondents were using GO (question 7); other frequently mentioned resources included EntrezGene, ChEBI, PSI-MOD, UniProt and the Plant Ontology. Interestingly, a number of groups were doing phenotype or anatomy, but each group was using a species-specific vocabulary.
There was strong interest for using text mining tools in batch processing mode (25 out of 28 respondents said that they would use this feature moderately/frequently/all the time—question 10). However, 22 out of 25 respondents also said that they would use interactive tools moderately or more frequently.
Discussion
Adaptation
Biocuration workflows have important commonalities and differences. Commonalities include document triage or prioritization, extraction and linkage of important biological entities, and extraction of relations and the underlying evidence for the relations. However, despite these commonalities, each biocuration workflow is different—in its inventory of biological entities, in its designation of what is ‘curation-relevant’, in the way that articles are prioritized for curation (by journal, by gene or protein, by novelty, etc.) and in how the workflow is divided among curators. Curators expressed a need for tools that could be easily adapted to the specific needs of their workflow and database, such as extensible lexicons that could be edited to include new relevant terms or to exclude terminological resources not relevant to the task. Another need was for tools that could tag the database-specific inventory of biological entities and relations, including numerical descriptions and parameters such as kinetic information.
If adaptation is needed, then a key question is: who is responsible for doing the adaptation—the tool developer or the curation team? Adaptation is a complex process and requires well-engineered, well-documented software, as well as sophisticated users/developers on the curation team. As mentioned above, it may require the construction of new lexicons and synonym lists, the writing of new hand-crafted patterns (for rule-based systems) and—for machine learning based systems—the ‘training’ of the system on application specific training data. Acquiring such training data and doing the training requires familiarity with annotation tools as well as experience in machine learning-based systems for natural language applications. A developer supporting a specific curated database may not have the time or expertise required to adapt natural language processing tools to the specific needs of their database.
Site-specific adaptation would also require each database to maintain its own curation pipeline and associated software. This could make it more difficult for curated databases to leverage ‘general purpose’ tools and could ultimately slow progress by making it more laborious to incorporate new tools or to upgrade the pipeline supporting the curation workflow. Due to these issues, adaptation requires close cooperation between the tool development team and the adopters of the tool.
One adaptation success story is Textpresso (6)—a tool that has been widely used across a number of databases; its website (www.textpressso.org) lists six ‘production sites’ and five additional pilot sites associated primarily with model organism databases. Textpresso grew out of the curation community and was developed to address needs of WormBase (www.wormbase.org). Since its initial deployment on WormBase, Textpresso developers have provided support for the porting of the tools to new application domains, working closely with the curators. Textpresso provides indexing of text (including full-text articles and pdfs) using a broad set of terms organized into biological categories, including incorporation of terms from existing ontologies, such as the Gene Ontology. The curator or other end user can compose a query by specifying combinations or patterns of indexed terms. This allows individual users to formulate queries to perform their custom tasks.
Literature access
Literature access is still a stumbling block for both biocurators and text mining developers. As noted above, curators need access to the full articles, including figures, tables and supplementary materials. Many research groups developing text mining tools have focused on abstracts, because these are easily accessible and can be downloaded as ASCII or XML. In contrast, access to full journal articles is complicated by difficulties in handling pdf and obtaining xml versions of the articles, as well as intellectual property issues. Although there are an increasing number of open access publications, curation teams need access to all of the relevant literature, not just to those journals that are more easily accessible.
What curators want from text mining tools
Through interviews, presentations at the workshop and the follow-up post-workshop survey, we have identified some curator desiderata. Curators wanted tools that were easy to use, easy to install and easy to maintain by the intended end user (ideally, a developer associated with the curation team, who will not necessarily be an expert in text mining or natural language processing). The tools do not have to be perfect, but they need to complement (not replace) the biocurator's function. A number of curation groups indicated that they would use the tools to do an initial batch processing, followed by biocurator validation, where the biocurator makes a yes/no decision and avoids having to type or look names up in a large database. Another important use was linking mentions of biological entities in text with the correct identifiers in biological databases, as well as linkage to the appropriate ontology terms. A number of curators felt that they would like text mining tools to aid in identifying and prioritizing papers for curation, to avoid wasting time on papers that did not have ‘relevant’ (e.g. curatable or novel) results. They also wanted tools to identify the sections of full-text papers containing curatable information.
Biocurators were also concerned about interoperability and data exchange, including formats that could communicate with other bioinformatics resources, either through the use of Web services, or via links to external resources and databases. Curators were interested in using text mining tools that could produce confidence scores, linkage to evidence passages in the text and ranking of automatically generated results, together with visualization aids, such as a customizable color-coding scheme for highlighting different levels annotations contained in a given article under curation.
What text mining developers need from curators
The biocurator community can assist by providing formalized descriptions of their workflows. Findings from our initial workflow studies indicated that each database may have a unique workflow—since databases typically differ in their criteria for what gets curated and in what order they do the various steps. Instrumentation of the curation interfaces would make it possible to gather data from curators on timing, throughput and patterns of use. This, in turn, would help to identify the major ‘choke points’ in the workflow. Based on such a workflow description and associated data on patterns of usage, the curators could work with the tool developers to identify appropriate insertion points for text mining in the workflow.
From the text mining tool developer point of view, it would be useful to have curators provide a more detailed description (and examples) of data selected as relevant and data designated as nonrelevant during the curation process. If annotations were saved on textual data that had been manually reviewed but deemed not curation relevant, this could serve as negative training data, crucial for the development and evaluation of text mining applications. It would also support comparison of current database content and automatically extracted annotations.
Conclusions and next steps
The biocuration community has an urgent need to ‘break the curation bottleneck’. Text mining tools have now progressed to the point where they can be useful to support expert biocurators—if inserted at the right points in the workflow, with the appropriate functionality and easy-to-use, easy-to-customize interfaces. A survey of biocurators revealed that two-thirds of the respondents had experimented with text mining, and over half were using some text mining tools in their workflow. The workshop on ‘Text Mining for the BioCuration Workflow’ at the third International Biocuration Conference (Berlin, 2009) represented an important step in opening a dialog between biological database curators and text mining developers. By continuing the conversation among the biocuration community, the bio-text mining researchers and the publishers, through workshops and challenge evaluations, we expect to see significant progress in this critical area.
There is now substantial momentum behind these interactions. Since the workshop in the spring of 2009, there have been two additional evaluations that have continued the exploration of these issues, with a third workshop planned for April 2012 and BioCreative IV planned for spring 2013.
BioCreative II.5 (11) (October 2009) compared curation of FEBS Letters articles on protein–protein interaction by authors, expert biocurators and automated systems. This work was inspired in part by community discussions around structured digital abstracts and the feasibility of author curation (19,20). The evaluation was organized with active participation of FEBS Letters, including both the editor (Gianni Cesareni) and the publisher (Elsevier), as well as a number of authors who participated in the author curation experiment. Two findings of relevance were that (i) authors had particular difficulty with the protein normalization step (the assignment of an appropriate UniProt identifier to a protein described in the article) and (ii) a post hoc combination of author plus automated system outperformed either one individually—in part, because the authors and the automated systems made very different kinds of mistakes. These results suggest that existing automated systems may be good enough now to help authors link genes or proteins mentioned in an article to the correct unique identifier; this might be a good candidate insertion point that could save time even for an experienced biocurator.
BioCreative III was held in September 2010, introducing a new ‘Interactive Annotation Task’, inspired in part by the findings from the April 2009 workshop. This interactive task focused on identifying which genes were being studied in an article and linking those genes to standard database identifiers. The task was designated as a demonstration task, with the goal of laying the groundwork for a rigorous evaluation of an interactive system for BioCreative IV (planned for spring 2013). To provide input from the end user and biocurator perspective, a User Advisory Group was organized to assess the six participating interactive systems and provided detailed feedback to the developer teams (12).
In addition to the above activities, the dialog is broadening to include the scientific publishing community, which is becoming an increasingly active partner. Both BioCreative II.5 and BioCreative III had active participation from the publisher community, and the Intelligent Systems for Molecular Biology (ISMB) conference has held successful sessions on Scientific Publishing for the last 3 years. However, the importance of this topic is not confined to the text mining, biocuration and scientific publishing communities. The maintenance of timely, high-quality computable resources provided by the growing number of curated databases derived from the scientific literature is critical to the entire scientific enterprise.
A direct follow on to the April 2009 Biocuration workshop will be held in association with the fifth International Biocuration Conference (spring 2012). This is organized as a BioCreative Satellite Workshop, with organizers including biological curators (Wu, Arighi from PRO and UniProt; Mattingly and Wiegers from the Comparative Toxicogenomic Database). The workshop will consist of three Tracks: Triage (Track 1): a collaborative biocuration-text mining development task for document prioritization for curation; Biocuration Workflows (Track 2): a collection of detailed descriptions of biocuration workflows and identification of insertion points for text mining, from the perspective of biocurators; and Interactive Text Mining (Track 3): an interactive text mining and user evaluation task, with evaluation by biocurators. Each of these tracks will have 6–9 participating groups.
The spring 2012 workshop described above will set the stage for BioCreative IV, to be held in the spring of 2013. We believe that these activities are greatly increasing communication among the diverse communities involved in biocuration. This, in turn, will lead to improved tools inserted into the biocuration workflow—driven by the needs and the insights of the biocurators.
Supplementary Data
Supplementary Data are available at Database Online.
Funding
National Science Foundation (grant IIS-0844419 to L.H.); US National Institutes of Health National Library of Medicine (grant 1G08LM10720-01 to C.N.A. and C. H. W.); Work related to BioCreative III was supported by the US National Science Foundation (grant DBI-0850319 to C.N.A., L.H., C.H.W.); the US National Institute of General Medical Sciences (grant R01-GM083871 to G.A.P.C.B.); the National Science Foundation (DBI-0849977 to G.A.P.G.B); the European Union Seventh Framework MICROME project (Grant Agreement Number 222886-2 to M.K. and A.V.); the US National Science Foundation IGERT (Grant 0221625 to K.G.D) and a PhRMA Foundation predoctoral fellowship in informatics; US National Science Foundation (grant DBI-0850219 to E.H.); US National Human Genome Research Institute (grant HG001315 to R.N.); National Institutes of Health (NIH) (grant 2U01HG02712-04 to A.L.V.) and European Commission contract FELICS (grant 021902RII3); National Institute of Environmental Health Sciences (NIEHS) and the National Library of Medicine (NLM) (R01ES014065 to T.W.); NIEHS (R01ES014065-04S1 to T.W.); National Institutes of Health National Center for Research Resources(P20RR016463 to T.W.); Biotechnology and Biological Sciences Research Council of the UK (grant BB/F010486/1 to A.G.W); the National Institutes of Health National Center for Research Resources (1R01RR024031 to A.G.W); the European Commission FP7 Program (2007-223411 to A.G.W). Funding for open access charge: The MITRE Corporation.
Conflict of interest. None declared.
Acknowledgements
We gratefully acknowledge the participation of Carl Schmidt and Peter D’Eustachio who contributed to the pre-workshop study of the biocuration workflow and Pascale Gaudet who helped on the survey for biocuration workflow and text mining. We also thank the anonymous reviewers for their critical observations, insights and suggestions. L.H. was responsible for the initial workshop organization and for the final editing of the article; G.A.P.C.B. interviewed several model organism databases and did the modeling of the workflows in UML, presented these at the workshop and contributed to Supplementary Appendix A1; M.K. interviewed BioGrid and MINT biocurators for their workflows, presented these at the workshop, and with A.V., created Table 2 and contributed to Supplementary Appendix A1; C.A. interviewed PRO and Reactome biocurators, ran the post-workshop survey and contributed the write-up in Supplementary Appendix B1; K.B.C. interviewed the CTD database and presented an overview of text mining at the workshop; C.H.W. contributed to workshop organization; the remaining authors (A.C.A., K.G.D., E.H., A.L., R.N., T.W., A.W., A.L.V.) provided inputs to the biocuration workflow study and contributed to Appendices A and B; all authors contributed to the writing of the article.
References
- 1.Galperin MY, Cochrane GR. The 2011 Nucleic Acids Research Database Issue and the online Molecular Biology Database Collection. Nucleic Acids Res. 2011;39(Suppl 1):D1–D6. doi: 10.1093/nar/gkq1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lanzen A, Oinn T. The Taverna Interaction Service: enabling manual interaction in workflows. Bioinformatics. 2008;24:1118–1120. doi: 10.1093/bioinformatics/btn082. [DOI] [PubMed] [Google Scholar]
- 3.Hull D, Wolstencroft K, Stevens R, et al. Taverna: a tool for building and running workflows of services. Nucleic Acids Res. 2006;34(Web Server issue):W729–W732. doi: 10.1093/nar/gkl320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burns GAPC, Krallinger M, Cohen KB, et al. Biocuration Workflow Catalogue—Text Mining for the Biocuration Workflow. 2009. Nature Precedings. http://dx.doi.org/10.1038/npre.2009.3250.1 (2 March 2012, date last accessed) [Google Scholar]
- 5.Krallinger M. A Framework for BioCuration Workflows (part II) Nature Precedings. 2009 http://dx.doi.org/10.1038/npre.2009.3126.1 (2 March 2012, date last accessed) [Google Scholar]
- 6.Muller HM, Kenny EE, Sternberg PW. Textpresso: an ontology-based information retrieval and extraction system for biological literature. PLoS Biol. 2004;2:e309. doi: 10.1371/journal.pbio.0020309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiegers TC, Davis AP, Cohen KB, et al. Text mining and manual curation of chemical-gene-disease networks for the comparative toxicogenomics database (CTD) BMC Bioinformatics. 2009;10:326. doi: 10.1186/1471-2105-10-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang P, Morgan AA, Zhang Q, et al. Automating document classification for the Immune Epitope Database. BMC Bioinformatics. 2007;8:269. doi: 10.1186/1471-2105-8-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fink JL, Kushch S, Williams PR, Bourne PE. BioLit: integrating biological literature with databases. Nucleic Acids Res. 2008;36:W385–W389. doi: 10.1093/nar/gkn317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prlic A, Martinez MA, Dimitropoulos D, et al. Integration of open access literature into the RCSB Protein Data Bank using BioLit. BMC Bioinformatics. 2010;11:220. doi: 10.1186/1471-2105-11-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leitner F, Chatr-aryamontri A, Mardis S, et al. The FEBS Letters/BioCreative II.5 experiment: making biological information accessible. Nat. Biotechnol. 2010;28:897–899. doi: 10.1038/nbt0910-897. [DOI] [PubMed] [Google Scholar]
- 12.Arighi C, Roberts P, Argawal S, et al. BioCreative III Interactive Task: an Overview. BMC Bioinformatics. 2011;12(Suppl 8):S1. doi: 10.1186/1471-2105-12-S8-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krallinger M, Leitner F, Rodriguez-Penagos C, Valencia A. Overview of the protein-protein interaction annotation extraction task of BioCreative II. Genome Biol. 2008;9(Suppl 2):S4. doi: 10.1186/gb-2008-9-s2-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lourenço A, Carneiro S, Carreira R, et al. Bringing Text Miners and Biologists Closer Together. Nature Precedings. 2009 http://dx.doi.org/10.1038/npre.2009.3188.1 (2 March 2012, date last accessed) [Google Scholar]
- 15.Dowell KG, McAndrews-Hill MS, Hill DP, et al. Integrating text mining into the MGI biocuration workflow. Database. 2009;2009:bap019. doi: 10.1093/database/bap019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veuthey A-L, Pillet V, Yip YL, Ruch P. Text mining for Swiss-Prot curation: A story of success and failure. Nature Precedings. 2009 http://dx.doi.org/10.1038/npre.2009.3166.1 (2 March 2012, date last accessed) [Google Scholar]
- 17.Ceol A, Chatr-Aryamontri A, Licata L, Cesareni G. Linking entries in protein interaction database to structured text: the FEBS Letters experiment. FEBS Lett. 2008;582:1171–1177. doi: 10.1016/j.febslet.2008.02.071. [DOI] [PubMed] [Google Scholar]
- 18.Bada M, Hunter L. Using the Gene Ontology to Annotate Biomedical Journal Articles. Nature Precedings. 2009 http://dx.doi.org/10.1038/npre.2009.3556.1 (2 March 2012, date last accessed) [Google Scholar]
- 19.Gerstein M, Seringhaus M, Fields S. Structured digital abstract makes text mining easy. Nature. 2007;447:142. doi: 10.1038/447142a. [DOI] [PubMed] [Google Scholar]
- 20.Hahn U, Wermter J, Blasczyk R, Horn PA. Text mining: powering the database revolution. Nature. 2007;448:130. doi: 10.1038/448130b. [DOI] [PubMed] [Google Scholar]