

Summary
Local Ca2+ transfer between adjoining domains of the sarcoendoplasmic reticulum (ER/SR) and mitochondria allows ER/SR Ca2+ release to activate mitochondrial Ca2+ uptake and to evoke a matrix [Ca2+] ([Ca2+]m) rise. [Ca2+]m exerts control on several steps of energy metabolism to synchronize ATP generation with cell function. However, calcium signal propagation to the mitochondria may also ignite a cell death program through opening of the permeability transition pore (PTP). This occurs when the Ca2+ release from the ER/SR is enhanced or is coincident with sensitization of the PTP. Recent studies have shown that several pro-apoptotic factors, including members of the Bcl-2 family proteins and reactive oxygen species (ROS) regulate the Ca2+ sensitivity of both the Ca2+ release channels in the ER and the PTP in the mitochondria. To test the relevance of the mitochondrial Ca2+ accumulation in various apoptotic paradigms, methods are available for buffering of [Ca2+], for dissipation of the driving force of the mitochondrial Ca2+ uptake and for inhibition of the mitochondrial Ca2+ transport mechanisms. However, in intact cells, the efficacy and the specificity of these approaches have to be established. Here we discuss mechanisms that recruit the mitochondrial calcium signal to a pro-apoptotic cascade and the approaches available for assessment of the relevance of the mitochondrial Ca2+ handling in apoptosis. We also present a systematic evaluation of the effect of ruthenium red and Ru360, two inhibitors of mitochondrial Ca2+ uptake on cytosolic [Ca2+] and [Ca2+]m in intact cultured cells.
Keywords: calcium, Ca2+, IP3 receptor, ryanodine receptor, mitochondria, VDAC, ruthenium red, Ru360
Mitochondrial Ca2+ transport mechanisms
The pathways of the mitochondrial Ca2+ import and export are illustrated in Fig1. Ca2+ traverses the outer mitochondrial membrane (OMM) primarily through the voltage dependent anion-selective channel (VDAC) [1-3]. The molecular nature of the proteins mediating the Ca2+ transport across the inner mitochondrial membrane (IMM) remains unknown. The protein mediating Ca2+ uptake is referred as the uniporter (UP) and has been identified as a Ca2+ selective ion channel [4]. The UP passes Ca2+ along the electrochemical gradient largely due to the highly negative mitochondrial membrane potential, ΔΨm of 180mV. Both the VDAC and the UP show Ca2+-dependent activation that is relevant for the homeostatic control of cytoplasmic [Ca2+] ([Ca2+]c) [1, 3, 4]. Ca2+ entering the mitochondrial matrix stimulates the Ca2+ sensitive mitochondrial dehydrogenases (CSMDH) to increase the H+ extrusion that is important for both the maintenance of the driving force for the Ca2+ uptake and for the ATP production. Ca2+ export across the OMM is mediated by the Ca2+ exchangers (Na+/Ca2+ and H+/Ca2+) and under some conditions (see below), the matrix Ca2+ induces formation of the PTP that traverses both the IMM and OMM and allows free passage of Ca2+, other ions and small molecules. Detailed discussion of the mitochondrial Ca2+ transport is available in recent comprehensive reviews [5-8].
Figure 1. Mechanisms of the mitochondrial Ca2+ transport.
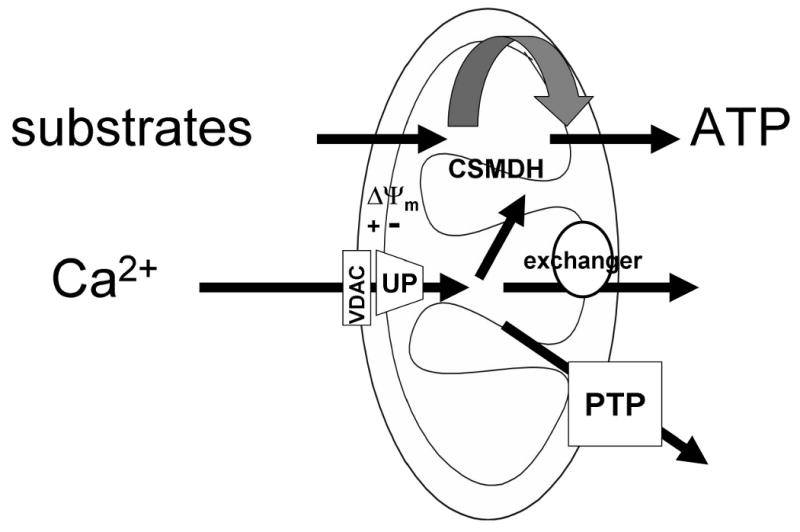
Abbreviations used: CSMDH, Ca2+ sensitive mitochondrial dehydrogenase; PTP, permeability transition pore; UP, uniporter; VDAC, voltage dependent anion selective channel.
Induction of cell death as a consequence of mitochondrial Ca2+ uptake
It has been known for several decades that sequestration of vast amounts of Ca2+ in the mitochondria occurs under various pathophysiological conditions and contributes to the demise of the cells [9]. In these paradigms, the loss of the balance between plasma membrane Ca2+ influx and Ca2+ export leads to a sustained elevation in [Ca2+]c from 100 nM to ≥ 1 μM, inducing a progressive increase in mitochondrial Ca2+ uptake. When large quantities of Ca2+ are accumulated in the mitochondrial matrix, Ca2+ interacts with cyclophilin D to induce opening of the PTP [10]. Furthermore, the rise in [Ca2+]m stimulates the generation of factors, including ROS and free fatty acids, which also promote the opening of the PTP [11, 12]. Opening of the PTP causes dissipation of the ΔΨm and release of Ca2+. If the cytoplasmic Ca2+ overload persists, the PTP stays open and allows accumulation of solutes in the mitochondrial matrix. This in turn, leads to expansion of the matrix space and to rupture of the OMM, giving rise to release of the intermembrane space content [13](Fig2A). Finally, impairment of the mitochondrial function and activation of cytoplasmic mechanisms by the released mitochondrial factors leads to execution of the cells.
Figure 2. Ca2+-induced mitochondrial membrane permeabilization.

Schemes illustrating possible mechanisms for the cellular Ca2+ overload (A)- and ER/SR Ca2+ mobilization (B,C) induced mitochondrial membrane permeabilization. Pro-survival mechanisms are depicted in green and pro-death mechanisms are shown in purple. Abbreviations used: AA, arachidonic acid; ROS, reactive oxygen species; cyto c, cytochrome c.
In the late 90s, studies of the propagation of the IP3 receptor (IP3R)- and ryanodine receptor (RyR)-mediated [Ca2+]c oscillations to the mitochondria provided the unexpected result that physiological [Ca2+]m transients can also trigger mitochondrial membrane permeabilization, leading to apoptotic cell death [14-16]. This pathway also depends on Ca2+-induced activation of the PTP. However, opening of the PTP in this case, required the coincidental detection of the [Ca2+]m oscillations and pro-apoptotic stimuli that lowered the threshold for the Ca2+-induced PTP opening (Fig2B). Neither the [Ca2+]m oscillations nor the sensitizing factor alone was sufficient to initiate these events. The PTP opening was followed by the release of pro-apoptotic factors from the intermembrane space to the cytoplasm. The exact mechanism of the OMM permeabilization remains elusive. An important attribute of this path is the absence of a prolonged and massive [Ca2+]c load. Therefore closure of the PTP may occur, preventing large scale swelling and allowing functional recovery of the mitochondria. Mitochondrial ATP production provides ATP for caspase activation enhancing the fidelity of the execution of the apoptotic program. Opening of the PTP by Ca2+ is effectively modulated by several factors, including ROS, pH, ΔΨm [5] the level of which is affected by a variety of metabolic intermediates, signalling molecules and drugs. Therefore, it is not surprising that the role of a “coincidence detection” or a “two hit” mechanism of Ca2+-dependent mitochondrial membrane permeabilization has been implicated in a broad range of cell death and tissue injury (e.g. [17]).
Recent studies of the ER-mitochondrial communication have also provided evidence for regulation of the IP3R-mediated Ca2+ release by both pro- and anti-apoptotic mechanisms (Fig2C). Pro-apoptotic factors have been reported to facilitate the ER Ca2+ mobilization and in turn, to increase the calcium signal propagation to the mitochondria, resulting in Ca2+-dependent mitochondrial membrane permeabilization. Due to a complex, local interaction between IP3Rs and mitochondrial Ca2+ uptake [18-20], regulation of ER Ca2+ release evokes more extensive changes in the [Ca2+]m signal than in the [Ca2+]c signal [21]. Boehning and co-workers showed high affinity binding of cytochrome c to the IP3Rs, which blocks the Ca2+-dependent inhibition of the IP3-induced Ca2+ efflux [22]. By this mechanism, cytochrome c released from the mitochondria at the onset of apoptosis recruits the IP3-linked Ca2+ mobilization from the ER and Ca2+ loading to the mitochondria to enhance and propagate the mitochondrial membrane permeabilization. A similar mechanism may be supported by the ROS that are produced and released from the mitochondria to the cytosol during mitochondrial membrane permeabilization [23] and promote the IP3R-mediated Ca2+ release [24]. The studies of Scorrano and co-workers provided evidence that the presence of Bax and Bak in the ER membrane is required to optimize the storage and mobilization of Ca2+ and showed that this effect is the critical contribution of Bax/Bak in several models of apoptosis [25]. The effect of Bax/Bak may be due to suppression of the interaction between the IP3R and the anti-apoptotic Bcl-2 [26, 27] or Bcl-xL [28], which controls the Ca2+ leakage from the ER. The recruitment of Bax to the ER and the ER Ca2+ release are also regulated by Bik, a BH3-only Bcl-2 family protein[29]. Bcl-2/Bcl-xL seems to serve both as a direct effector of the IP3R [26, 28] and as a regulator of the phosphorylation of the type 1 IP3R [27]. Thus the effect of Bcl-2/Bcl-xL on the activation of the IP3R by IP3 [26, 28] may reflect more than one mechanism of the interaction or interaction with more than one IP3R isotype and may be modulated by numerous factors that target the IP3R.
In the sequence from the opening of the IP3Rs to the opening of the PTP, the calcium signal propagation may be affected by several factors, including the activity of the mitochondrial Ca2+ uptake sites, the organization of the ER-mitochondrial interface, and the morphology of the mitochondria. tBid, a pro-apoptotic BH3 only Bcl-2 family protein induces permeabilization of the OMM to promote the propagation of the IP3R-mediated [Ca2+]c signal to the mitochondria [30]. Overexpression of Mcl-1, an anti-apoptotic Bcl-2 family protein [31] or PPARgamma-coactivator-1alpha (PGC-1α) that triggers mitochondrial biogenesis [32] has been shown to lower the efficacy of the mitochondrial Ca2+ uptake, the IP3R-mediated [Ca2+]m signal and the Ca2+-dependent apoptosis. Neutralization or down-regulation of these factors may favor to an increase in the apoptotic potential of the [Ca2+]m signal. Regarding the role of the interface, we observed that the ER-mitochondrial association becomes more extensive in the beginning of apoptosis induced by tunicamycin or serum starvation. Furthermore, when strengthening of the associations was induced by expression of a synthetic bi-functional ER-mitochondrial crosslinker, enhanced Ca2+ transfer from ER to the mitochondria and sensitization to the Ca2+-dependent mitochondrial membrane permeabilization were noted (Csordas, Renken, Varnai, Balla, Mannella and Hajnoczky, unpublished). The ER-mitochondrial interface may be supported by direct links between the organelles or by anchorage of the organelles to the cytoskeletal framework. Both the ER and mitochondria display dynamic positioning along the cytoskeletal tracks and through the control of the organellar motility the local Ca2+ signalling between the organelles is also modulated [33, 34]. The fusion and fission dynamics of the mitochondria emerges as an important factor for the organization of the ER-mitochondrial interface as well as for the distribution of Ca2+ accumulated to the mitochondrial matrix. Szabadkai et al. showed that Drp1 mediated fragmentation of the mitochondria inhibited the spreading of the [Ca2+]m signal along the organelles [35]. These authors also found that mitochondrial fragmentation suppressed the ceramide-induced apoptosis that is mediated through Ca2+ delivery to the mitochondria. Thus evidence is on the rise that some determinants of the local Ca2+ transfer between ER and mitochondria and of the mitochondrial morphology may also serve as checkpoints for the calcium signal to support either cell survival or cell death.
Methods for evaluating the relevance of the mitochondrial handling in the induction of apoptosis
The emerging evidence for the calcium signal-driven mitochondrial membrane permeabilization underscores the need for methods to test the dependence of various apoptosis paradigms on mitochondrial Ca2+ uptake. The commonly used strategies include (1) buffering of [Ca2+]c, (2) dissipation of the driving force of the mitochondrial Ca2+ uptake and (3) inhibition of the mitochondrial uptake sites (Fig3). For each strategy, fairly straightforward protocols have been established in suspensions of isolated mitochondria. However, in intact cells, the barrier formed by the plasma membrane, the complex cellular response to mitochondrial inhibition and the extra-mitochondrial targets of the drugs limit the utility of these protocols.
Figure 3. Approaches for preventing the mitochondrial Ca2+ uptake in intact cells.

(1) Buffering of [Ca2+]c
Introduction of Ca2+ chelators into the cytoplasm provides a means to attenuate the [Ca2+]c signal. BAPTA or EGTA can be loaded to the cells as acetoxymethylesters that are cleaved by intracellular esterases to release the free acid form. Alternatively, BAPTA or EGTA free acids can be introduced to the cells by microinjection. To assess the effect of the chelator on the [Ca2+]c fluorescence measurement of [Ca2+]c by a cytoplasmic probe (e.g. fura2, fluo4 or cytoplasm targeted pericam) can be used. It is important to note that effective buffering of bulk [Ca2+]c does not necessarily result in prevention of the [Ca2+]m signal. In fact demonstration of the IP3-linked [Ca2+]m signal in the absence of a [Ca2+]c rise in Ca2+ chelator loaded cells has served as a major evidence for the local Ca2+ transfer between the IP3R and adjacent mitochondrial Ca2+ uptake sites [19, 20]. However, ≥ 1mM BAPTA was sufficient to also eliminate the IP3R-mediated [Ca2+]m signal [20]. Notably, in myocytes and myotubes, that have rigorously controlled cytoarchitecture, Ca2+ transfer from RyR to the mitochondria was shown even in the presence of 1-5mM BAPTA[36, 37].
(2) Dissipation of the driving force of the mitochondrial Ca2+ uptake
Inhibitors of respiratory chain and protonophores (uncouplers) can be used to collapse the ΔΨm, the primary component of the driving force of the Ca2+ uptake. Rotenone, a complex I inhibitor and antimycin A or myxothiazol, complex III inhibitors are applied both in combination and separately. A potential drawback of these protocols is the increase in mitochondrial ROS generation. Carbonylcyanide p-trifluoromethoxyphenylhydrazone (FCCP) or carbonylcyanide m-chlorophenylhydrazone (CCCP) are the most commonly used protonophores. The effect of these agents on ΔΨm can be monitored by fluorescent membrane potential probes like tetramethylrhodamine methyl ester (TMRM) or JC1. Notably, in cells that display a high plasma membrane potential, a considerable component of the TMRM or JC1 accumulation reflects the plasma membrane potential [38]. In most cell types, antimycin A or FCCP/CCCP represent effective means to dissipate the ΔΨm. A problem is that the uncoupled mitochondria do not only stop producing ATP but consume ATP through the reverse operation of the F1F0 ATPase. To slow down cellular ATP depletion, the respiratory chain inhibitors and uncouplers are commonly added together with oligomycin, a blocker of the F1F0 ATPase. This combination may allow for maintaining the ATP levels in cells where mitochondria have only a minor role in producing cellular ATP.
(3) Inhibition of the mitochondrial uptake sites
For inhibition of the Ca2+ uniporter and the VDAC, ruthenium derivates, Ruthenium Red (RuRed) and Ru360 have been used. In isolated mitochondria, RuRed is a potent inhibitor of the mitochondrial Ca2+ uptake and has no major considerable side effects. Based on its highly charged nature, RuRed would not be expected to traverse the plasma membrane. Still, RuRed was reported to enter isolated cardiomyocytes [39] and has been used recently to inhibit the VDAC and the uniporter in many cell types. However, the effect of RuRed on mitochondrial Ca2+ uptake was not validated in most studies presumably, because of the difficulty in monitoring [Ca2+]m in intact cells. In regard to the specificity of the inhibition of the VDAC and UP it is relevant that RuRed also inhibits plasma membrane and intracellular cation channels (e.g. TASK-3[40], TRPV [41] and RyR[42], respectively).
To evaluate the utility of RuRed in inhibition of mitochondrial Ca2+ uptake in intact cells, we recorded [Ca2+]m simultaneously with [Ca2+]c in 3 cell lines, RBL-2H3 rat mast cells, HepG2 human hepatoma cells and H9c2 rat cardiac myoblast cells using a mitochondrial matrix-targeted pericam construct [43] and cytosolic fura2 (Fig4). The cells were pretreated 12-18h with RuRed of 5 or 10μM, a supramaximal dose for the inhibition of the mitochondrial Ca2+ uptake in permeabilized cells or in isolated mitochondria. In the single cell [Ca2+] imaging experiments, the cells were first incubated in a Ca2+ free medium and were sequentially stimulated by an IP3-linked agonist and thapsigargin to mobilize Ca2+ from the ER. Then, extracellular Ca2+ was added back to allow the Ca2+ entry (basal+store-depletion induced). RBL-2H3 cells expressing the M1 muscarinic receptor displayed a robust carbachol-induced [Ca2+]c transient that was associated with a sharp and large increase in [Ca2+]m (Fig4A). The Ca2+ entry elicited a substantial increase in [Ca2+]c, which was followed by a further rise in [Ca2+]m. RuRed did not cause any change in either the [Ca2+]c or [Ca2+]m signal evoked by the intracellular Ca2+ mobilization or Ca2+ entry (Fig4A red traces). In HepG2 cells, the agonist-induced [Ca2+]c and [Ca2+]m increase was relatively small, whereas the Ca2+ entry resulted in a large elevation in both [Ca2+]c and [Ca2+]m (Fig4B). Similar to the RBL-2H3 cells, RuRed failed to suppress either the [Ca2+]c or the [Ca2+]m signal in the HepG2 cells (Fig4B, red traces). Previously, RuRed was shown to inhibit IP3 and the Ca2+-induced [Ca2+]m signal in permeabilized cells [20]. Therefore, the lack of an effect in the intact cells seems to indicate that RuRed did not enter the RBL-2H3 or the HepG2 cells.
Figure 4. RuRed-and Ru360-dependent changes in calcium signalling in intact and permeabilized cells.
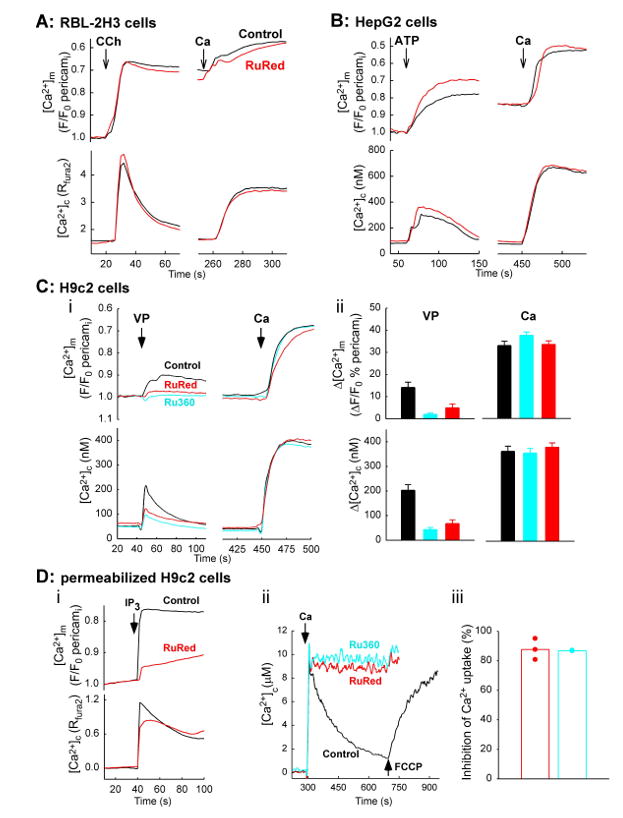
Cells cultures were transfected with mitochondria targeted inverse pericam (24h) and subsequently, pretreated with RuRed (Fluka, ≥ 85% purity, red) or Ru360 (Calbiochem, cyan) for 12-18h in serum-free medium: RBL-2H3, RuRed 5μM (A); HepG2, RuRed 10μM (B); H9c2, RuRed or Ru360 10μM each. Subsequently, cells were loaded with fura2/AM and incubated in a Ca2+ free extracellular medium. For the RuRed/Ru360 treated cells the drug was also present during the fura2 loading and the recording. Fluorescence [Ca2+]c and [Ca2+]m imaging was carried out as described before [3, 20]. Traces represent the means for 3-7 experiments for each condition.
(A) The RBL-2H3 cells were co-transfected with pericam and M1 muscarinic receptor. Cells were treated with carbachol, CCH of 100μM, thapsigargin, Tg of 2μM and CaCl2, Ca of 2mM as indicated by the arrows.
(B) HepG2 cells were treated with ATP 100μM, Tg of 2μM and CaCl2 of 5mM.
(C) H9c2 cells were treated with vasopressin, VP of 100nM, Tg of 2μM and CaCl2 of 10mM (i). Bar charts show the size of the VP- and Ca-induced [Ca2+]m and [Ca2+]m rise calculated in single cells (ii). The rates for the Ca-induced [Ca2+]c rise were 31.3±4.1 nM/s; 29.4±2.7 nM/s; 23.6±4 nM/s and for the [Ca2+]m rise were 2.4±0.4 %/s; 2.6±0.5 %/s; 2.6±0.3 %/s in control, RuRed-and Ru360-pretreated cells, respectively (n=34, 30 and 32 cells).
(D) RuRed-and Ru360 induced suppression of the mitochondrial Ca2+ uptake in permeabilized H9c2 cells
(i) Pericam-transfected adherent H9c2 cells were permeabilized with saponin and incubated in an intracellular medium containing fura2 5μM. [Ca2+]c and [Ca2+]m were followed in single cells with CCD imaging of fura2 and pericam fluorescence, respectively. To evoke a Ca2+ release from the ER IP3 7.5μM was added. RuRed of 5μM was added 3min before IP3 (red trace). Traces are means of three experiments.
(ii,iii) Suspensions of H9c2 cells (1mg protein/ml) were incubated in an intracellular medium and were permeabilized by digitonin. [Ca2+]c was monitored in a fluorometer using fura2FF (0.5μM) added to the bathing medium. CaCl2 30μM and FCCP of 5μM was added as indicated by the arrows. RuRed (red) and Ru360 (blue) 1μM each was added 1min before Ca2+ addition. Representative traces for the fluorometer records (ii) and bar charts showing the RuRed/Ru360 sensitive fraction of the decay in [Ca2+]c (iii).
A more complex picture emerged in the H9c2 cells. RuRed caused attenuation of the agonist-induced [Ca2+]c signal, indicating an effect of the drug on the cascade from the agonist receptor (V1 vasopressin receptor) to the ER Ca2+ mobilization (Fig4Ci, red vs. black). Accordingly, the agonist-induced [Ca2+]m signal was also suppressed in the H9c2 cells. The Ca2+ entry resulted in a [Ca2+]c signal that was followed by a rapid and large [Ca2+]m elevation both in the control and RuRed-treated H9c2 cells. RuRed did not affect the rate or the extent of the [Ca2+]m rise. Thus RuRed failed to inhibit the mitochondrial Ca2+ uptake in the H9c2 cells. Ru360 has been reported as a more potent inhibitor of the mitochondrial Ca2+ uptake than RuRed, which lacks the inhibitory effect of RuRed on the RyR and enters rapidly intact cells [44]. Exposure of the H9c2 cells to Ru360 (10μM for 12-24h) led to some attenuation of the agonist-induced [Ca2+]c and [Ca2+]m response and no change occurred in the Ca2+ entry dependent [Ca2+]c and [Ca2+]m rise. Since the individual H9c2 cells show some heterogeneity in the [Ca2+]c and [Ca2+]m and a modest drug-induced change in the underlying Ca2+ fluxes may remain unnoticed in the cell population average, the size and the rate for the agonist and Ca2+-induced [Ca2+]c and [Ca2+]m signal was also quantitated in single cells (Fig4Cii). However, the single cell analysis did not show any effects of RuRed or Ru360 on the [Ca2+]m rise. Thus, similar to RuRed, Ru360 also failed to inhibit the mitochondrial Ca2+ uptake in intact H9c2 cells. In permeabilized H9c2 cells (Fig4D), the IP3-induced Ca2+ mobilization appeared as a sharp [Ca2+]c increase closely followed by an increase in [Ca2+]m (Fig4Di). When RuRed was present the IP3-induced [Ca2+]c rise was somewhat suppressed but the [Ca2+]m increase was almost abolished (Fig4Di red). Furthermore, when Ca2+ was added to permeabilized H9c2 cell suspensions the mitochondrial Ca2+ uptake led to a rapid and uncoupler-sensitive decay in [Ca2+]c, which was practically abolished by both RuRed and Ru360 (Fig4Dii,iii). These data confirmed that RuRed or Ru360 is a potent inhibitor of the mitochondrial Ca2+ uptake in permeabilized H9c2. Taken together the results, neither RuRed nor Ru360 interferes with the [Ca2+]c signal propagation to the mitochondria in intact H9c2 cells, presumably due to the limited penetration across the plasma membrane. Furthermore, both RuRed and Ru360 exert an inhibition in the agonist-induced Ca2+ mobilization. The main conclusion is that the extracellular application of neither RuRed nor Ru360 can be used in intact RBL-2H3, HepG2 or H9c2 cells for evaluating the role of mitochondrial Ca2+ uptake in cell death. For broader implications, it seems to be necessary to validate the effect on mitochondrial Ca2+ uptake when RuRed or Ru360 is intended to be used in any intact cells.
Complementary approaches to test the role of mitochondrial Ca2+ in cell death also use inhibitors of the PTP opening. Two commonly used cell-permeable inhibitors are cyclosporin A and bongkrekic acid, which bind to cyclophilin D and to the adenine nucleotide translocase, respectively. Importantly, cyclosporin A does not abolish the opening of the pore, it only causes an increase in the Ca2+ retention capacity of the mitochondria. The broad extramitochondrial effects of cyclosporin A, including the inhibition of calcineurin and the impairments of the mitochondrial ATP production evoked by bongkrekic acid represent concerns regarding the prolonged treatment of intact cells. However, in the absence of molecular approaches, cyclosporin A (or the N-methyl-4-valine-cyclosporin that inhibits the PTP without calcineurin inactivation [45]) and bongkrekic acid particularly, in combination with approaches to suppress the mitochondrial Ca2+ uptake, have been proven practical for testing the relevance of the mitochondrial calcium PTP pathway in induction of cell death.
Conclusions
Decoding of calcium signals in the mitochondria to cell death inducing mechanisms depends on coincidental detection of a [Ca2+]m rise and pro-apoptotic stimuli. In the calcium signal propagation from the ER to the mitochondria, basically every step offers an opportunity for other signalling mechanisms to alter the spatial and temporal properties of the [Ca2+]m signal. Furthermore, a variety of pro-apoptotic factors converge on the PTP to control its Ca2+ sensitivity. To sort out the pathophysiological relevance of these mechanisms the mitochondrial Ca2+ uptake has to be targeted. The drugs available for the inhibition of mitochondrial Ca2+ uptake have limitations and may evoke complex cellular responses. At least, the monitoring of [Ca2+]m in intact cells has become feasible by the recent addition of a series of luminescent or green fluorescent protein-based fluorescent probes and fluorophores.[43, 46-48]. Evaluation of the efficacy and specificity of the available protocols may provide an adequate solution until the molecular structure of the mitochondrial Ca2+ transporters will be defined and new potent inhibitors will be synthetized.
Acknowledgments
This work was supported by grants from the NIH to GH and an AHA Scientist Development Grant 0435236N to GC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gincel D, Zaid H, Shoshan-Barmatz V. Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem J. 2001;358:147–55. doi: 10.1042/0264-6021:3580147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, Tuft RA, Fogarty KE, Rizzuto R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol. 2002;159:613–24. doi: 10.1083/jcb.200205091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bathori G, Csordas G, Garcia-Perez C, Davies E, Hajnoczky G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and VDAC. J Biol Chem. 2006 April 5; doi: 10.1074/jbc.M600906200. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 4.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–4. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 5.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–55. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 6.Pfeiffer DR, Gunter TE, Eliseev R, Broekemeier KM, Gunter KK. Release of Ca2+ from mitochondria via the saturable mechanisms and the permeability transition. IUBMB Life. 2001;52:205–12. doi: 10.1080/15216540152846019. [DOI] [PubMed] [Google Scholar]
- 7.Carafoli E. Historical review: mitochondria and calcium: ups and downs of an unusual relationship. Trends Biochem Sci. 2003;28:175–81. doi: 10.1016/S0968-0004(03)00053-7. [DOI] [PubMed] [Google Scholar]
- 8.Nicholls DG, Chalmers S. The integration of mitochondrial calcium transport and storage. J Bioenerg Biomembr. 2004;36:277–81. doi: 10.1023/B:JOBB.0000041753.52832.f3. [DOI] [PubMed] [Google Scholar]
- 9.Krieger C, Duchen MR. Mitochondria, Ca2+ and neurodegenerative disease. Eur J Pharmacol. 2002;447:177–88. doi: 10.1016/s0014-2999(02)01842-3. [DOI] [PubMed] [Google Scholar]
- 10.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–61. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 11.Starkov AA, Chinopoulos C, Fiskum G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium. 2004;36:257–64. doi: 10.1016/j.ceca.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 12.Scorrano L, Penzo D, Petronilli V, Pagano F, Bernardi P. Arachidonic acid causes cell death through the mitochondrial permeability transition. Implications for tumor necrosis factor-alpha aopototic signaling. J Biol Chem. 2001;276:12035–40. doi: 10.1074/jbc.M010603200. [DOI] [PubMed] [Google Scholar]
- 13.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–9. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 14.Szalai G, Krishnamurthy R, Hajnoczky G. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. Embo J. 1999;18:6349–61. doi: 10.1093/emboj/18.22.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. Embo J. 2001;20:2690–701. doi: 10.1093/emboj/20.11.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pacher P, Hajnoczky G. Propagation of the apoptotic signal by mitochondrial waves. Embo J. 2001;20:4107–21. doi: 10.1093/emboj/20.15.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacobson J, Duchen MR. Mitochondrial oxidative stress and cell death in astrocytes--requirement for stored Ca2+ and sustained opening of the permeability transition pore. J Cell Sci. 2002;115:1175–88. doi: 10.1242/jcs.115.6.1175. [DOI] [PubMed] [Google Scholar]
- 18.Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 1993;262:744–7. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- 19.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280:1763–6. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 20.Csordas G, Thomas AP, Hajnoczky G. Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. Embo J. 1999;18:96–108. doi: 10.1093/emboj/18.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin X, Varnai P, Csordas G, Balla A, Nagai T, Miyawaki A, Balla T, Hajnoczky G. Control of calcium signal propagation to the mitochondria by inositol 1,4,5-trisphosphate-binding proteins. J Biol Chem. 2005;280:12820–32. doi: 10.1074/jbc.M411591200. [DOI] [PubMed] [Google Scholar]
- 22.Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5:1051–61. doi: 10.1038/ncb1063. [DOI] [PubMed] [Google Scholar]
- 23.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–14. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng Y, Shen X. H2O2 directly activates inositol 1,4,5-trisphosphate receptors in endothelial cells. Redox Rep. 2005;10:29–36. doi: 10.1179/135100005X21660. [DOI] [PubMed] [Google Scholar]
- 25.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–9. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 26.Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol. 2004;166:193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci U S A. 2005;102:105–10. doi: 10.1073/pnas.0408352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK. The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol. 2005;7:1021–8. doi: 10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mathai JP, Germain M, Shore GC. BH3-only BIK regulates BAX,BAK-dependent release of Ca2+ from endoplasmic reticulum stores and mitochondrial apoptosis during stress-induced cell death. J Biol Chem. 2005;280:23829–36. doi: 10.1074/jbc.M500800200. [DOI] [PubMed] [Google Scholar]
- 30.Csordas G, Madesh M, Antonsson B, Hajnoczky G. tcBid promotes Ca(2+) signal propagation to the mitochondria: control of Ca(2+) permeation through the outer mitochondrial membrane. Embo J. 2002;21:2198–206. doi: 10.1093/emboj/21.9.2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Minagawa N, Kruglov EA, Dranoff JA, Robert ME, Gores GJ, Nathanson MH. The anti-apoptotic protein Mcl-1 inhibits mitochondrial Ca2+ signals. J Biol Chem. 2005;280:33637–44. doi: 10.1074/jbc.M503210200. [DOI] [PubMed] [Google Scholar]
- 32.Bianchi K, Vandecasteele G, Carli C, Romagnoli A, Szabadkai G, Rizzuto R. Regulation of Ca2+ signalling and Ca2+-mediated cell death by the transcriptional coactivator PGC-1alpha. Cell Death Differ. 2006;13:586–96. doi: 10.1038/sj.cdd.4401784. [DOI] [PubMed] [Google Scholar]
- 33.Yi M, Weaver D, Hajnoczky G. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol. 2004;167:661–72. doi: 10.1083/jcb.200406038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brough D, Schell MJ, Irvine RF. Agonist-induced regulation of mitochondrial and endoplasmic reticulum motility. Biochem J. 2005;392:291–7. doi: 10.1042/BJ20050738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16:59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 36.Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu SS. Transport of Ca2+ from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J Bioenerg Biomembr. 2000;32:97–104. doi: 10.1023/a:1005520714221. [DOI] [PubMed] [Google Scholar]
- 37.Shkryl VM, Shirokova N. Transfer and tunneling of Ca2+ from sarcoplasmic reticulum to mitochondria in skeletal muscle. J Biol Chem. 2006;281:1547–54. doi: 10.1074/jbc.M505024200. [DOI] [PubMed] [Google Scholar]
- 38.Nicholls DG. Simultaneous Monitoring of Ionophore- and Inhibitor-mediated Plasma and Mitochondrial Membrane Potential Changes in Cultured Neurons. J Biol Chem. 2006;281:14864–74. doi: 10.1074/jbc.M510916200. [DOI] [PubMed] [Google Scholar]
- 39.McCormack JG, England PJ. Ruthenium Red inhibits the activation of pyruvate dehydrogenase caused by positive inotropic agents in the perfused rat heart. Biochem J. 1983;214:581–5. doi: 10.1042/bj2140581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Czirjak G, Enyedi P. Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J Biol Chem. 2002;277:5426–32. doi: 10.1074/jbc.M107138200. [DOI] [PubMed] [Google Scholar]
- 41.Amann R, Maggi CA. Ruthenium red as a capsaicin antagonist. Life Sci. 1991;49:849–56. doi: 10.1016/0024-3205(91)90169-c. [DOI] [PubMed] [Google Scholar]
- 42.Hymel L, Schindler H, Inui M, Fleischer S. Reconstitution of purified cardiac muscle calcium release channel (ryanodine receptor) in planar bilayers. Biochem Biophys Res Commun. 1988;152:308–14. doi: 10.1016/s0006-291x(88)80715-0. [DOI] [PubMed] [Google Scholar]
- 43.Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+ Proc Natl Acad Sci U S A. 2001;98:3197–202. doi: 10.1073/pnas.051636098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, Phillips R, Altschuld R, Katsube Y, Sperelakis N, Bers DM. Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J Biol Chem. 1998;273:10223–31. doi: 10.1074/jbc.273.17.10223. [DOI] [PubMed] [Google Scholar]
- 45.Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol. 2002;62:22–9. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
- 46.Rudolf R, Mongillo M, Rizzuto R, Pozzan T. Looking forward to seeing calcium. Nat Rev Mol Cell Biol. 2003;4:579–86. doi: 10.1038/nrm1153. [DOI] [PubMed] [Google Scholar]
- 47.Gerasimenko O, Tepikin A. How to measure Ca2+ in cellular organelles? Cell Calcium. 2005;38:201–11. doi: 10.1016/j.ceca.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 48.Demaurex N. Calcium measurements in organelles with Ca2+-sensitive fluorescent proteins. Cell Calcium. 2005;38:213–22. doi: 10.1016/j.ceca.2005.06.026. [DOI] [PubMed] [Google Scholar]