

Abstract
Exposure of the fetus to excess estrogen is believed to increase the risk of developing breast cancer during adult life. Fetal exposure to low doses of the xenoestrogen bisphenol A resulted in long-lasting effects in the mouse mammary gland that were manifested during adult life. It enhanced sensitivity to estradiol, decreased apoptosis, increased the number of progesterone receptor-positive epithelial cells at puberty and increased lateral branching at 4 months of age. We now report that fetal exposure to 2.5, 25, 250 and 1000μg bisphenol A/kg body weight/day induces the development of ductal hyperplasias and carcinoma in situ at postnatal day 50 and 95 in rats. These highly proliferative lesions have an increased number of estrogen receptor-α positive cells. Thus, fetal bisphenol A exposure is sufficient to induce the development of preneoplastic and neoplastic lesions in the mammary gland in the absence of any additional treatment aimed at increasing tumor development.
Keywords: Bisphenol A, xenoestrogen, mammary gland, rat, carcinogenesis, carcinoma in situ
Introduction
Recent data have suggested that perturbations in the fetal environment may predispose individuals to disease and/or organ dysfunction, which become apparent in adulthood [1,2]. This new emphasis on the fetal origins of adult diseases has prompted scientists to hypothesize that fetal exposure to environmental estrogens may be the underlying cause of the increased incidence of uterine leiomyoma, testicular cancer and breast cancer observed in European and US populations over the last 50 years [3–5].
Epidemiological studies suggest that fluctuating estrogen levels in the fetal environment have long-term consequences regarding the risk of developing breast cancer during adult life [6–8]. Given the long latency period between exposure and effect, epidemiological studies designed to explore this hypothesis have used prenatal and perinatal markers of in utero estrogen exposure because direct estrogen measurements are not available from birth records. For instance, twin pregnancy was used as an indicator of high estrogen exposure and pre-eclampsia for low estrogen exposure. Increased risk of breast cancer correlated with twin dizygotic birth and pre-eclampsia was associated with lowered risk in several studies [7,9].
Direct evidence of prenatal estrogen exposure and breast cancer risk is being gathered from the cohort of women born to mothers treated with diethylstilbestrol (DES) during pregnancy. This potent synthetic estrogen was administered to women as an anti-abortive therapy between the years 1948 and 1971 in the US, Europe and Australia. Tragically, this therapy was continued long after it was shown to be ineffective, and was finally stopped in the early 1970’s when a rare pathology, clear cell adenocarcinoma of the vagina [10,11], as well as other abnormalities of the uterus, oviduct and cervix were diagnosed in young women that had been exposed to DES in utero [12,13]. These women are now reaching the age at which breast cancer becomes more prevalent. It has been reported that in the group of women exposed in utero to DES, aged 40 years and older, there is a 2.5 fold increase in the incidence of breast cancer compared to unexposed women of the same age [14], suggesting that indeed, prenatal exposure to synthetic estrogens may play an important role in the development of breast neoplasms.
Consistent with this observation, experiments in rats showed that prenatal exposure to DES resulted in increased mammary cancer incidence during adulthood [15]. These experiments illustrated that rats exposed prenatally to DES and challenged with the chemical carcinogen dimethylbenzanthracene (DMBA) at puberty had a significantly greater incidence of palpable mammary tumors at 10 months of age than animals exposed prenatally to vehicle. In addition, the tumor latency period was shorter in the DES-exposed compared to the vehicle-exposed group. Both the epidemiological and experimental data are consistent with the hypothesis that excessive estrogen exposure during development may increase the risk of developing breast cancer.
In utero exposure to tamoxifen, an estrogen antagonist and partial agonist, has also been shown to increase the incidence of mammary tumors when the exposed offspring are challenged with DMBA at puberty. Eighteen weeks after the challenge, 95% of the tamoxifen-exposed animals developed tumors compared to 50% of the vehicle-treated rats [16]. However, in the above-mentioned studies, both DES and tamoxifen were administered at high pharmacological doses to reflect the medical use of these agents, while the effects of twinning and pre-eclampsia represent a physiological range of endogenous hormone levels to which developing fetuses are exposed. Yet there is a third type of exposure that needs to be addressed, i.e. the inadvertent and continuous exposure of fetuses to environmentally active xenoestrogens.
Among these compounds, bisphenol A (BPA) is receiving increased attention due to its ubiquitous presence in the environment and chronic human exposure. BPA is used in the manufacture of polycarbonate plastics and epoxy resins, and leaches from food containers [17], beverage containers [18] and dental sealants and composites [19] under normal conditions of use [5]. BPA has been measured in maternal and fetal plasma and placental tissue at birth in humans [20,21]. A recent study of 394 Americans reported that BPA was found in 95% of urine samples [22]. From these data, the mean exposure was estimated to be 40ng/kg body weight (BW)/day and the 95th percentile was 230ng/kg BW/day assuming that 70% of the daily dose was excreted into the urine. A smaller study reported a mean daily urinary excretion of BPA at levels of 1.2μg and estimated the maximum daily intake of BPA to be 0.23μg/kg BW [23]. Alternatively, daily intake can be estimated from the amount of BPA leached from food containers, beverage containers and dental materials. Using this approach, a probable exposure range of 2–20μg BPA/kg BW/day was calculated [24]. It is worth noting that the US Environmental Protection Agency estimates the lowest observable adverse effects level to be 50mg BPA/kg BW/day and from this the safe dose was calculated to be 50μg BPA/kg BW/day [25].
Exposure of rodents to low doses of BPA during fetal development has been shown to alter a variety of biological endpoints including early vaginal opening [26], early onset of puberty [27], disrupted estrous cyclicity [28,29], and decreased levels of luteinizing hormone following ovariectomy [29].
Our previous work has focused on the effects of perinatal BPA exposure on mouse mammary gland development. In these mice, BPA caused a decreased invasion of the stromal compartment, increased number of terminal end buds (TEBs) relative to the ductal area, decreased apoptosis and increased numbers of cells expressing progesterone receptor in the pubertal mammary gland [30]. At 4 months of age, these animals had a significant increase in lateral branching [30]. By 6 months of age, we observed an overall increase in epithelial structures including terminal ends and a premature appearance of alveolar buds, normally associated with pregnancy in the mouse [31]. More importantly, BPA exposed mice that were ovariectomized prepubertally showed an enhanced sensitivity to estradiol demonstrated by an increase in the number of TEBs, TEB area, TEB density and ductal extension [30,32]. It has also been observed that increased ductal density, estimated from mammographic density, is associated with increased risk for developing breast cancer [33]. Based on all these findings, we hypothesize that perinatal exposure to environmental levels of BPA increases the risk of developing mammary cancer. To explore this hypothesis, we have chosen to use a rat model because it mimics the human disease regarding hormone factors and histopathology more closely [34,35] than the available mouse models [36].
In the rat model, treatment with chemical carcinogens such as DMBA and N-nitrosomethylurea (NMU) results in the development of intraductal hyperplasias, intraductal carcinomas in situ (CIS) and adenocarcinomas. Intraductal hyperplasias are believed to be the precursor lesion for both CIS and adenocarcinomas [37]. Additionally, transplantation studies have revealed that intraductal hyperplasias give rise to palpable tumors [38], and thus are considered preneoplastic lesions. The aim of the present work was to examine whether fetal BPA exposure is sufficient to induce the development of preneoplastic lesions in the mammary gland in the absence of any additional treatment aimed at increasing tumor development (i.e., chemical carcinogen, pharmacological hormone treatment, etc.).
Materials and Methods
Fetal exposure to BPA
Sexually mature female Wistar-Furth rats (8 week-old; Harlan, Indianapolis, IN) were maintained in temperature-controlled and light-controlled (14-h light, 10-h dark cycle) conditions in the Tufts University School of Medicine animal facility. All experimental procedures were approved by the Tufts University–New England Medical Center Animal Research Committee in accordance with the Guide for Care and Use of Laboratory Animals. Cages and bedding tested negligible for estrogenicity by the E-SCREEN assay [39]; water was supplied from glass bottles only. Food (Harlan Teklad 2018) was supplied ad libitum. Estrogenicity of the feed was measured at 20 femtomoles of estrogen equivalents per gram, a negligible amount [39]. Female rats were mated with Wistar-Furth males of proven fertility and the morning on which sperm was observed in vaginal smears was designated embryonic day (E)1. On E9, the rats were weighed and implanted with Alzet osmotic pumps (Alza Corp., Mountain View, CA) designed to deliver the following doses of BPA/kg BW/day: 2.5μg, 25μg, 250μg or 1000μg. For convenience, these will be subsequently referred to as BPA2.5, BPA25, BPA250 and BPA1000, respectively. The control animals were implanted with a pump delivering 50% dimethyl sulfoxide (vehicle control; Sigma Chemical Co., St Louis, MO). Thus, the fetuses were exposed to BPA or vehicle from E9 until postnatal day (PND) 1. A wide range of BPA doses were tested because, contrary to the mouse model, to our knowledge there are no data available on developmental effects of BPA in the mammary gland of rats, with the exception of one study on Sprague-Dawley rats exposed to 0.1 and 1.2mg BPA/kg BW/day from E5 to weaning via drinking water [29].
Parameters of growth and sexual maturation
The number of live and dead pups was recorded on the day of delivery. On PND 2, the sex of each offspring was recorded and when necessary, litters were culled to a maximum of 8 pups. On PND 4, the anogenital distance (AGD) of each pup was measured. Whole litters were weighed on PND 4, 7 and 11; hence the body weight data collected before weaning (PND 21) was not segregated by sex. After weaning, individual weights were recorded for female pups at regular intervals until PND 110. Beginning on PND 28, female offspring were checked daily for vaginal opening.
Mammary gland collection
Female offspring from all treatment groups were sacrificed either at PND 50, during puberty when evident ductal growth and stroma invasion was underway, or at PND 95, when ducts have extended towards the edge of the fat pad. The 4th and 5th left inguinal mammary glands were fixed and processed for paraffin embedding and the contralateral glands were whole-mounted.
Whole mounts and histology
The tissue was fixed overnight in 10% phosphate buffered formalin and processed for paraffin embedding as described previously [40]. Sections (5μm) were stained with hematoxylin and eosin (H&E).
The whole mounts were prepared following protocols described previously [41]. Briefly, the mammary glands were removed and spread on a 75 × 50 × 1 mm glass slide (Fisher Scientific, Pittsburgh, PA), fixed overnight in 10% phosphate buffered formalin, after which they were dehydrated in alcohol, cleared of fat with toluene, rehydrated and stained with carmine alum. After staining, the whole mounts were dehydrated as described above, cleared in xylene, and bagged in Kpak® SealPak heat-seal pouches (Kpak Corp., Minneapolis, MN) with methyl salicylate.
Morphometric analysis
The whole mounts were viewed with a stereomicroscope Stemi 2000 (Carl Zeiss, Munchen-Hallbergmoos, Germany) and the histological sections were visualized with an Axioskop 2 Plus microscope (Carl Zeiss). Images were captured with an AxioCam HR color digital camera (Carl Zeiss) and the Axiovision software (version 4.3).
Three 5μm sections separated by 50μm were used to assess the presence of pre-neoplastic lesions in the mammary glands of PND 50 and PND 95 females. The leading edge and TEBs were localized and starting 400μm from the most proximal TEB, a 4mm2 box was drawn; all the ducts located within this area were counted (Supplementary Figure 1) and classified according to Singh et al [34]. The main criterion used to diagnose a ductal hyperplasia was an increase in the number of epithelial cells lining the ducts (3–4 cells thick). The incidence of preneoplastic lesions was expressed in comparison to the total number of ductal structures present in the 4mm2 area analyzed. The diagnosis of CIS was performed following the criteria described by Russo et al [42] and Singh et al [34].
Immunohistochemistry
An antigen retrieval method using microwave pretreatment and 0.01 M sodium citrate buffer (pH 6) was performed as previously described [43]. A monoclonal mouse antibody for estrogen receptor α (ERα, Novocastra, Newcastle, UK) and rabbit polyclonal anti-Ki67 (Vector Laboratories, Burlingame, CA) were used at 1:150 and 1:3000 dilutions, respectively. The antigen-antibody reaction was visualized using the streptavidin-peroxidase complex, with diaminobenzidine tetrahydrochloride (Sigma-Aldrich) as the chromogen. Counterstaining was performed with Harris’ hematoxylin and the sections were imaged as described above. One 5μm section from the mammary gland of each animal was used to assess the presence of Ki67 and ERα immunostaining in PND 50 and PND 95 females. For each section, a 1mm2 box was drawn at a distance 400μm from the most proximal TEB and all the ductal structures located within this box were analyzed. The ducts were classified as normal or hyperplastic (including CIS) and the numbers of positive and negative cells were recorded for Ki67 or ERα for each structure.
Statistics
As individual offspring cannot be exposed prenatally, we exposed the dams. Hence, we were careful to randomize maternal effects and maximize the number of maternal units represented in each group. For each histological measurement, only one individual from a given litter was assigned to each group and end point. All calculated parameters and statistical significance were determined using SPSS statistical software (Chicago, IL). Overall differences in parameters were analyzed by ANOVA. When significant, post-hoc tests (Bonferroni or planned t-tests) were used to make comparisons between treatment groups. For all statistical tests, results were considered significant at p<0.05. All results are presented as mean +/− standard error of the mean (SEM).
Results
Growth and reproductive parameters
No statistically significant differences in body weight were observed between the females born to BPA- and vehicle-exposed dams at any point between PND 4–110 (Supplementary Figure 2). Similarly, no differences were observed regarding age at vaginal opening, litter size, or sex ratios among the treatment groups (Table 1). However, male offspring in the BPA250 group had a significant reduction in AGD when compared to males born to control and BPA2.5 dams, whereas AGD was not affected by BPA treatment in the female offspring (Table 1).
Table 1.
Reproductive parameters of control and BPA offspring
| No. of live newborns | Sex ratio, % litter female | Age at vaginal opening (days) | Female AGD on PND 4 (mm) | Male AGD on PND 4 (mm) | |
|---|---|---|---|---|---|
| Control | 6.10 ± 0.50 | 51.0 ± 5.28 | 34.56 ± 0.481 | 2.05 ± 0.071 | 4.10 ± 0.056 a |
| BPA 2.5 | 6.95 ± 0.44 | 52.2 ± 3.39 | 34.89 ± 0.317 | 2.09 ± 0.048 | 4.03 ± 0.074 b |
| BPA25 | 8.09 ± 0.84 | 56.4 ± 6.50 | 34.17 ± 0.292 | 2.13 ± 0.097 | 4.00 ± 0.109 |
| BPA250 | 7.20 ± 0.62 | 58.2 ± 5.45 | 34.12 ± 0.294 | 1.93 ± 0.070 | 3.59 ± 0.098 ab |
| BPA1000 | 7.18 ± 0.54 | 54.2 ± 5.40 | 34.88 ± 0.399 | 1.98 ± 0.072 | 3.86 ± 0.303 |
Common superscripts denote statistically significant differences (a, p<0.001; b, p=0.001).
BPA induces preneoplastic lesions in prenatally exposed mammary glands
Analysis of whole mounts revealed the presence of densely stained structures mostly in terminal ducts (Figure 1) consistent with the appearance of ductal hyperplasias. This diagnosis was confirmed by histological analysis of the microdissected areas (not shown).
Figure 1.
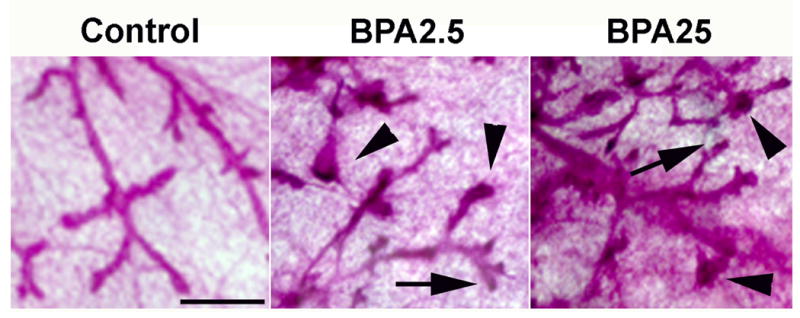
Mammary gland whole mounts at PND 95 from control, BPA2.5 and BPA25 groups. The BPA treated animals developed intraductal hyperplasias, mostly in the terminal ducts (arrowheads). The denser staining represents the abundance of epithelial cells compared to normal terminal ducts (arrows). Scale bar: 500μm.
Histological sections of the mammary glands were analyzed at PND 50 and PND 95. While ductal hyperplasias were observed in all animals analyzed, exposure of fetuses to BPA resulted in a significant 3–4 fold increase in the incidence of hyperplastic ducts relative to the controls at PND 50 (Figure 2A, p<0.05). This increase was quantitatively similar at all BPA doses. At PND 95 the percentage of hyperplastic ducts was lower overall than the PND 50 animals (Figure 2D). Interestingly, only the incidence of hyperplastic lesions observed in the BPA2.5 group was significantly higher than those of the vehicle-exposed controls (p=0.032).
Figure 2.
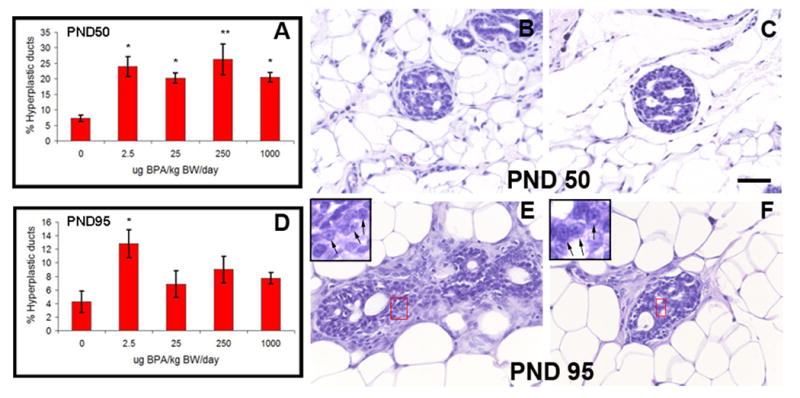
Neoplastic lesions. The percentage of ductal hyperplasias is significantly increased in BPA exposed animals at PND 50 (A) and PND 95 (D); *, p<0.05; **, p<0.005. Some of the ductal lesions were identified as CIS and had a cribriform pattern. This can be observed at PND 50 (B and C) and at PND 95 (E and F). The CIS showed not only multiple lumina but also hyperchromatic nuclear staining with visible nucleoli (inset in E and F). Scale bar: 50μm
In addition to the increased number of hyperplastic ducts, cribriform-like structures were also observed in the mammary glands of BPA250 and BPA1000 rats at PND 50 (Figure 2B–C) and PND 95 (Figure 2E–F). There was a 25% (BPA250: 1/4, BPA1000: 1/4) incidence in both BPA-treated groups at PND 50, while it reached 33% (BPA250: 2/6, BPA1000: 2/6) in both groups at PND 95. These structures were classified as CIS as they exhibited the hallmarks of these structures, namely (i) an increased ductal size due to proliferation of the luminal epithelial cells, (ii) enlargement of the luminal epithelial cells along with the presence of nucleoli and a variability in chromatin pattern and (iii) rounded luminal spaces (secondary lumina) formed by trabecular rods of cells aligned perpendicular to the longer axis of the duct.
Increased ERα and Ki67 expression in ductal hyperplasias
Ki67 immunostaining was performed in sections from PND 50 and PND 95 animals to verify that the epithelial cells in the hyperplasias and CIS were proliferating. Indeed, the number of Ki67-positive epithelial cells was increased in the ductal hyperplasias (developed in all animals regardless of the exposure) and CIS compared to the normal ducts, indicating an increased proliferative activity in the aberrant structures at PND 50 (Figure 3 A–B) and PND 95 (data not shown). Likewise, the hyperplastic lesions and CIS exhibited a higher percentage of ERα-positive cells than the normal ducts (Figure 3 C–D). Quantification showed that, after combining the data from all normal ducts and all hyperplastic lesions (regardless of dose), Ki67 and ERα expression were significantly increased in the lesions relative to normal ductal structures (Figure 3E; PND50: Ki67, p=0.009, n=19; ERα, p<0.001, n=18; PND95: Ki67 and ERα, p<0.001, n=26 for each marker).
Figure 3.
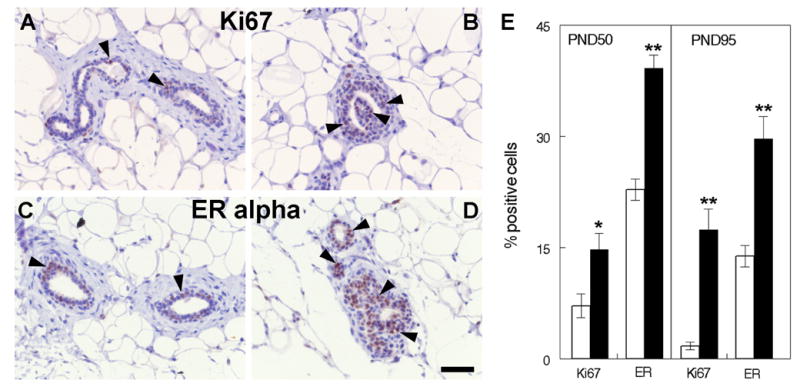
Ki67 and ERα expression in BPA exposed mammary glands. Ductal hyperplasias show an increased expression of Ki67 and ERα (B and D, arrowheads) compared to normal ducts (A and C). Representative pictures are from PND 50 animals. The bar graph depicts the percent of Ki67 and ERα positive cells at PND 50 and PND 95. White bars represent normal ducts, black bars represent neoplastic lesions (*, p<0.05; **, p<0.001 with respect to normal ducts). Scale bar: 50μm.
Discussion
The results described in this report provide compelling evidence that developmental exposure to BPA induces neoplastic transformation of the mammary gland, a concept inferred from our previously published work, but never before tested. Indeed, fetal exposure to BPA increased the number of intraductal hyperplasias in the mammary gland at all doses tested. The relevance of this finding is that intraductal hyperplasias are considered to be the precursors of carcinomas both in rodents and humans [34]. Moreover, intraductal hyperplasias have been shown to develop into palpable tumors when transplanted into cleared fat pads of hosts with intact ovaries [38]. Even more striking was the presence of CIS in the mammary glands of the animals exposed to the two higher BPA doses which were observed at puberty (PND 50) and at three months of age, the final time point of this study (PND 95). Whether the incidence of CIS might be increased in the BPA-exposed animals at later time points remains to be determined.
The appearance of these lesions at puberty is reminiscent of the timing of appearance of DES-induced clear cell carcinoma of the vagina in humans, which manifested with a peak incidence at 19 years of age, suggesting that exposure to ovarian hormones contribute to the development of these pathologies [13]. In the mammary gland, the peripubertal period is characterized by intense ductal morphogenesis encompassing tissue remodeling, epithelial invasion of the stroma, increased rates of cell proliferation and cell death; hence, the pubertal mammary gland is particularly prone to neoplastic development [30,44]. Indeed, an increased percentage of proliferating cells was observed in hyperplastic areas and CIS compared to the normal ducts. In addition, the percentage of ERα-positive cells was also increased in these lesions, suggesting that the proliferative activity in these lesions may be estrogen-mediated. One can speculate that these epithelial cells are already primed and will respond more strongly to the high levels of estradiol during puberty and adulthood. In this regard, it is worth noting that mammary carcinomas in both rats and humans are predominantly “estrogen-dependent”. Further, the peripubertal period is considered the most vulnerable regarding the sensitivity of the rat mammary gland to chemical carcinogenesis [44,45], and of the human mammary gland to irradiation [46].
A recent publication showed that BPA treatment induces proliferative pre-cancerous lesions in the rat prostate [47]. In this study, neonatal exposure to 10μg BPA/kg BW/day on PND 1, 3 and 5 did not increase the incidence of prostatic intraepithelial neoplasias (PINs). However, the increased propensity of the BPA-exposed animals to develop PINs was revealed when those rats were given a hormonal stimulus (testosterone plus estradiol for 16 weeks) during adulthood. Similarly, Durando et al showed that fetal exposure to 25μg BPA/kg BW/day induced the development of mammary gland neoplasia which was manifested after treatment with NMU during adulthood [48].
The present report as well as the above-mentioned publications show that developmental exposure to BPA enhances the likelihood of developing carcinomas in both the mammary gland and the prostate [47,48]. Further work will be necessary to verify whether the ductal hyperplasias and CIS we have observed in the mammary gland have the ability to generate invasive carcinomas. This possibility is currently being tested in our laboratory by treating animals exposed to BPA in utero with a subcarcinogenic dose of NMU at puberty [49].
The mechanisms underlying the origin and progression of neoplastic lesions after developmental exposure to natural or synthetic estrogens or xenoestrogens are still largely unknown. Supporters of the novel theory of fetal origins of adult disease are proposing that epigenetic changes such as DNA methylation and chromatin remodeling play a central role in carcinogenesis [50]. Ho and collaborators postulate that permanent alterations in the DNA methylation patterns of multiple cell signaling genes identified in the BPA-exposed prostates may be the underlying cause of neoplastic development later in life [47]. In contrast, most investigators in the field of cancer research support the idea that cancer is due to the accumulation of mutations in a cell (somatic mutation theory) [51]. Both the genetic and epigenetic theories of carcinogenesis imply that cancer originates in a cell that has undergone genetic and/or epigenetic changes, which ultimately result in dysregulated cell proliferation [52].
Alternatively, the tissue organization field theory postulates that carcinogenesis represents a problem of tissue organization, comparable to organogenesis gone awry, and that proliferation is the default state of all cells [53,54]. According to this theory, carcinogens and teratogens disrupt the normal dynamic interaction of neighboring cells and tissues during early development and/or adulthood [40,41]. As a result of the disruption in tissue organization, cells would regain their constitutive ability to proliferate and promote neoplastic development.
An example of tumor development likely due to the disruption of tissue organization is the DES-induced clear cell carcinoma of the vagina. This carcinoma originates from areas of cervicovaginal adenosis, which are regions of simple columnar epithelium that develop within the stratified squamous epithelium of the vagina [55,56]. The development of cervicovaginal adenosis has been attributed to aberrant cell-fate determination in which some vaginal epithelial cells acquire a uterine fate and become a simple columnar epithelium rather than a stratified squamous one [57]. The mesenchyme plays a major role in this epithelial fate determination process [58]. DES may also act directly on the vaginal epithelial cells by blocking the expression of p63, a protein that plays a major role in the fate determination of the vaginal and other stratified squamous epithelia [57]. More recently, msx2 which plays a critical role in cell fate determination in the vaginal epithelium, was shown to be repressed by DES [58]. This homeodomain transcription factor is required for the correct expression of wnt7a, and the absence of msx2 would result in a complete failure of stratification of the vaginal epithelium [59].
In contrast with the genital tract, very little is known about how estrogens affect mammary gland morphogenesis. Prenatal exposure of mice to 250ng BPA/kg BW/day resulted in advanced maturation of the fat pad at E18 [60]. Changes in the appearance of the mammary epithelium were also observed such as decreased cell size, delayed lumen formation, and increased ductal area. Because maturation of the fat pad is the driving event for ductal growth and branching, it is likely that the increased ductal area in BPA-exposed mice is due to the acceleration of this process.
Similar to the female genital tract, several members of the wnt signaling cascade and msx2 are expressed during fetal mammary gland development [61]. The expression of wnt4, wnt5b, and msx2 is regulated by estrogens in the adult mammary gland [62,63]. Hence, it is plausible that fetal xenoestrogen exposure may result in the extemporaneous expression of this set of genes that, in turn, may cause altered morphogenesis and neoplastic development as is the case for clear cell carcinoma of the vagina. In addition, it is also conceivable that fetal BPA exposure may result in alterations in the methylation patterns of genes involved in the reciprocal tissue interactions that mediate morphogenesis.
In summary, the results presented in this report buttress the link between fetal exposure to BPA and the development of neoplasias in the adult mammary gland. These neoplasias may have their origin in the altered morphogenesis that occurs in the fetus during the period of BPA exposure [64]. Finally, this study supports the hypothesis that environmental exposure to xenoestrogens during fetal life may contribute to the increased incidence of breast cancer observed over the past five decades.
Supplementary Material
Schematic representation of a 5μm mammary gland section. The morphometric analysis was performed in a 4mm2 area that excluded the TEBs. All structures within this area were counted and classified.
Body weights of offspring born to mothers exposed to BPA or vehicle. Mean body weights of male and female offspring (PND 4–11) and mean body weights of female offspring (PND 22–110) showed no statistically significant differences due to BPA treatment.
Acknowledgments
The authors are grateful to April Flynn for technical assistance, to Laura Vandenberg and Cheryl Schaeberle for their editorial assistance, and to Dr Beverly S Rubin for critical reading of the manuscript. This work was supported by NIH grants ES012301 and ES08314.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sallout B, Walker M. The fetal origin of adult diseases. Journal of Obstetrics and Gynaecology. 2003;23:555–60. doi: 10.1080/0144361031000156483. [DOI] [PubMed] [Google Scholar]
- 2.Barker DJP, Hanson MA. Altered regional blood flow in the fetus: the origins of cardiovascular disease? Acta Paediatricia. 2004;93:1559–60. [PubMed] [Google Scholar]
- 3.Sharpe RM, Skakkebaek NE. Are oestrogens involved in falling sperm count and disorders of the male reproductive tract? Lancet. 1993;341:1392–5. doi: 10.1016/0140-6736(93)90953-e. [DOI] [PubMed] [Google Scholar]
- 4.Skakkebaek NE, Meyts ER, Jorgensen N, et al. Germ cell cancer and disorders of spermatogenesis: an environmental connection? APMIS. 1998;106:3–12. doi: 10.1111/j.1699-0463.1998.tb01314.x. [DOI] [PubMed] [Google Scholar]
- 5.Markey CM, Rubin BS, Soto AM, Sonnenschein C. Endocrine disruptors from Wingspread to environmental developmental biology. J Steroid Biochem Molec Biol. 2003;83:235–44. doi: 10.1016/s0960-0760(02)00272-8. [DOI] [PubMed] [Google Scholar]
- 6.Trichopoulos D. Is breast cancer initiated in utero? Epidemiology. 1990;1:95–6. [PubMed] [Google Scholar]
- 7.Braun MM, Ahlbom A, Floderus B, Brinton LA, Hoover RN. Effect of twinship on incidence of cancer of the testis, breast, and other sites (Sweden) CCC. 1995;6:519–24. doi: 10.1007/BF00054160. [DOI] [PubMed] [Google Scholar]
- 8.Ekbom A, Trichopoulos D, Adami HO, Hsieh CC, Lan SJ. Evidence of prenatal influences on breast cancer risk. Lancet. 1992;340:1015–8. doi: 10.1016/0140-6736(92)93019-j. [DOI] [PubMed] [Google Scholar]
- 9.Potischman N, Troisi R. In-utero and early life exposures in relation to risk of breast cancer. CCC. 1999;10:561–73. doi: 10.1023/a:1008955110868. [DOI] [PubMed] [Google Scholar]
- 10.Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina: association of maternal stilbestrol therapy with tumor appearance in young women. New Engl J Med. 1971;284:878–81. doi: 10.1056/NEJM197104222841604. [DOI] [PubMed] [Google Scholar]
- 11.Mittendorf R. Teratogen update: carcinogenesis and teratogenesis associated with exposure to diethylstilbestrol (DES) in utero. Teratology. 1995;51:435–45. doi: 10.1002/tera.1420510609. [DOI] [PubMed] [Google Scholar]
- 12.Herbst AL, Bern HA. Developmental Effects of Diethylstilbestrol (DES) in Pregnancy. New York: Thieme-Stratton; 1988. [Google Scholar]
- 13.Herbst AL, Anderson D. Clear cell adenocarcinoma of the vagina and cervix secondary to intrauterine exposure to diethylstilbestrol. Seminars in Surgical Oncology. 1990;6:343–6. doi: 10.1002/ssu.2980060609. [DOI] [PubMed] [Google Scholar]
- 14.Palmer JR, Hatch EE, Rosenberg CL, et al. Risk of breast cancer in women exposed to diethylstilbestrol in utero: preliminary results (United States) CCC. 2002;13:753–8. doi: 10.1023/a:1020254711222. [DOI] [PubMed] [Google Scholar]
- 15.Boylan ES, Calhoon RE. Transplacental action of diethylstilbestrol on mammary carcinogenesis in female rats given one or two doses of 7,12-dimethylbenz(a)anthracene. Cancer Res. 1983;43:4879–84. [PubMed] [Google Scholar]
- 16.Hilakivi-Clarke L, Cho E, Onojafe I, Liao DJ, Clarke R. Maternal exposure to tamoxifen during pregnancy increases carcinogen-induced mammary tumorigenesis among female rat offspring. Clin Cancer Res. 2000;6:305–8. [PubMed] [Google Scholar]
- 17.Brotons JA, Olea-Serrano MF, Villalobos M, Olea N. Xenoestrogens released from lacquer coating in food cans. Environ Health Perspect. 1994;103:608–12. doi: 10.1289/ehp.95103608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biles JE, McNeal TP, Begley TH, Hollifield HC. Determination of Bisphenol-A in reusable polycarbonate food-contact plastics and migration to food simulating liquids. J Agric Food Chem. 1997;45:3541–4. [Google Scholar]
- 19.Olea N, Pulgar R, Perez P, et al. Estrogenicity of resin-based composites and sealants used in dentistry. Environ Health Perspect. 1996;104(3):298–305. doi: 10.1289/ehp.96104298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schonfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M, Chahoud I. Parent bisphenol A accumulation in the human maternal-fetal-placental unit. Environ Health Perspect. 2002;110:A703–A707. doi: 10.1289/ehp.110-1241091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y, Taketani Y. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum Reprod. 2002;17:2839–41. doi: 10.1093/humrep/17.11.2839. [DOI] [PubMed] [Google Scholar]
- 22.Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Ekong J, Needham JL. Urinary concentrations of bisphenol A and 4-nonylphenol in a human reference population. Environ Health Perspect. 2005;113:391–5. doi: 10.1289/ehp.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arakawa C, Fujimaki K, Yoshinaga J, Imai H, Serizawa S, Shiraishi H. Daily urinary excretion of bisphenol A. Environmental Health and Preventative Medicine. 2004;9:22–6. doi: 10.1265/ehpm.9.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagel SC, vom Saal FS, Thayer KA, Dhar MG, Boechler M, Welshons WV. Relative binding affinity-serum modified access (RBA-SMA) assay predicts the relative in vivo bioactivity of the xenoestrogens bisphenol A and octylphenol. Environ. Health Perspect. 1997;105:70–6. doi: 10.1289/ehp.9710570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bisphenol A. CASRN 80-05-7. 1993 http://www.epa.gov/iris/subst/0356.htm.
- 26.Honma S, Suzuki A, Buchanan DL, Katsu Y, Watanabe H, Iguchi T. Low dose effects of in utero exposure to bisphenol A and diethylstilbestrol on female mouse reproduction. Reproductive Toxicology. 2002;16:117–22. doi: 10.1016/s0890-6238(02)00006-0. [DOI] [PubMed] [Google Scholar]
- 27.Howdeshell KL, Hotchkiss AK, Thayer KA, Vandenbergh JG, vom Saal FS. Exposure to bisphenol A advances puberty. Nature. 1999;401:763–4. doi: 10.1038/44517. [DOI] [PubMed] [Google Scholar]
- 28.Markey CM, Coombs MA, Sonnenschein C, Soto AM. Mammalian development in a changing environment: exposure to endocrine disruptors reveals the developmental plasticity of steroid-hormone target organs. Evolution and Development. 2003;5:1–9. doi: 10.1046/j.1525-142x.2003.03011.x. [DOI] [PubMed] [Google Scholar]
- 29.Rubin BS, Murray MK, Damassa DA, King JC, Soto AM. Perinatal exposure to low doses of bisphenol-A affects body weight, patterns of estrous cyclicity and plasma LH levels. Environ Health Perspect. 2001;109:675–80. doi: 10.1289/ehp.01109675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munoz de Toro MM, Markey CM, Wadia PR, et al. Perinatal exposure to bisphenol A alters peripubertal mammary gland development in mice. Endocrinology. 2005;146:4138–47. doi: 10.1210/en.2005-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Markey CM, Luque EH, Munoz de Toro MM, Sonnenschein C, Soto AM. In utero exposure to bisphenol A alters the development and tissue organization of the mouse mammary gland. Biol Reprod. 2001;65:1215–23. doi: 10.1093/biolreprod/65.4.1215. [DOI] [PubMed] [Google Scholar]
- 32.Wadia PR, Vandenberg LN, Rubin BS, Sonnenschein C, Soto AM. Perinatal exposure to bisphenol A alters sensitivity of the mouse mammary gland to estradiol; Endocrinology Society Meeting; 2006. p. 78. [Google Scholar]
- 33.McCormack VA, Dos Santos Silva I. Breast density and parenchymal patterns as markers of breast cancer risk: a meta-analysis. Cancer Epidem Biomar. 2006;15:1159–69. doi: 10.1158/1055-9965.EPI-06-0034. [DOI] [PubMed] [Google Scholar]
- 34.Singh M, McGinley JN, Thompson HJ. A comparison of the histopathology of premalignant and malignant mammary gland lesions induced in sexually immature rats with those occurring in the human. Lab Invest. 2000;80:221–31. doi: 10.1038/labinvest.3780025. [DOI] [PubMed] [Google Scholar]
- 35.Thompson HJ, Singh M. Rat models of premalignant breast disease. J Mammary Gland Biol Neoplasia. 2000;5:409–20. doi: 10.1023/a:1009582012493. [DOI] [PubMed] [Google Scholar]
- 36.Nandi S, Guzman R, Yang J. Hormones and mammary carcinogenesis in mice, rats, and humans: a unifying hypothesis. Proc Nat Acad Sci USA. 1995;92:3650–7. doi: 10.1073/pnas.92.9.3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thompson HJ, McGinley JN, Wolfe P, Singh M, Steele VE, Kelloff GJ. Temporal sequence of mammary intraductal proliferations, ductal carcinomas in situ and adenocarcinomas induced by 1-methyl-1-nitrosourea in rats. Carcinogenesis. 1998;19:2181–5. doi: 10.1093/carcin/19.12.2181. [DOI] [PubMed] [Google Scholar]
- 38.Haslam SZ, Bern HA. Histopathogenesis of 7,12-diemthylbenz(a)anthracene-induced rat mammary tumors. Proc Nat Acad Sci USA. 1977;74:4020–4. doi: 10.1073/pnas.74.9.4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soto AM, Lin T-M, Justicia H, Silvia RM, Sonnenschein C. An “in culture” bioassay to assess the estrogenicity of xenobiotics. In: Colborn T, Clement C, editors. Chemically induced alterations in sexual development: the wildlife/human connection. Princeton: Princeton Scientific Publishing; 1992. pp. 295–309. [Google Scholar]
- 40.Maffini MV, Soto AM, Calabro JM, Ucci AA, Sonnenschein C. The stroma as a crucial target in rat mammary gland carcinogenesis. J Cell Sci. 2004;117:1495–502. doi: 10.1242/jcs.01000. [DOI] [PubMed] [Google Scholar]
- 41.Maffini MV, Calabro JM, Soto AM, Sonnenschein C. Stromal regulation of neoplastic development: Age-dependent normalization of neoplastic mammary cells by mammary stroma. Am J Pathol. 2005;67:1405–10. doi: 10.1016/S0002-9440(10)61227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Russo J, Russo IH, Rogers AE, Van Zwieten MJ, Gusterson BA. Tumours of the mammary gland. In: Turusov VS, Mohr U, editors. Pathology of tumours in laboratory animals Vol I Tumors of the rat. Lyon: IARC Scientific Publication N99; 1990. pp. 47–78. [PubMed] [Google Scholar]
- 43.Maffini MV, Ortega H, Stoker C, Giardina R, Luque EH, Munoz de Toro MM. Bcl-2 correlates with tumor ploidy and nuclear morphology in early stage prostate carcinoma. Path Res Pract. 2001;197:487–92. doi: 10.1078/0344-0338-00116. [DOI] [PubMed] [Google Scholar]
- 44.Russo IH, Russo J. Mammary gland neoplasia in long-term rodent studies. Environ Health Perspect. 1996;104:938–67. doi: 10.1289/ehp.96104938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grubbs CJ, Peckham JC, Cato KD. Mammary carcinogenesis in rats in relation to age at time of N-nitroso-N-methylurea administration. J Nat Cancer Inst. 1983;70:209–12. [PubMed] [Google Scholar]
- 46.Land CE, Tokunaga M, Koyama K, et al. Incidence of female breast cancer among atomic bomb survivors, Hiroshima and Nagasaki, 1950–1990. Radiation Research. 2003;160:707–17. doi: 10.1667/rr3082. [DOI] [PubMed] [Google Scholar]
- 47.Ho S-M, Tang WY, Belmonte de Frausto J, Prins GS. Developmental exposure to estradiol and bisphenol a increases susceptibility to prostate carcinogenesis and epigenetically regulates phosphodiesterase type 4 variant 4. Cancer Res. 2006;66:5624–32. doi: 10.1158/0008-5472.CAN-06-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Durando M, Kass L, Piva J, et al. Prenatal bisphenol A exposure induces preneoplastic lesions in the mammary gland in Wistar rats. Environ Health Perspect. 2006 doi: 10.1289/ehp.9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murray TJ, Maffini MV, Sonnenschein C, Munoz de Toro MM, Luque EH, Soto AM. Perinatal exposure to bisphenol A and mammary gland cancer; Endocrinology Society Meeting; 2006. p. 205. [Google Scholar]
- 50.Li S, Hursting SD, Davis BJ, McLachlan JA, Barrett JC. Environmental exposure, DNA methylation, and gene regulation: lessons from diethylstilbesterol-induced cancers. Ann NY Acad Sci. 2003;983:161–9. doi: 10.1111/j.1749-6632.2003.tb05971.x. [DOI] [PubMed] [Google Scholar]
- 51.Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331–42. doi: 10.1038/nrc795. [DOI] [PubMed] [Google Scholar]
- 52.Weinberg RA. The Biology of Cancer. New York: Taylor & Francis; 2006. [Google Scholar]
- 53.Soto AM, Sonnenschein C. The somatic mutation theory of cancer: growing problems with the paradigm? BioEssays. 2004;26:1097–107. doi: 10.1002/bies.20087. [DOI] [PubMed] [Google Scholar]
- 54.Sonnenschein C, Soto AM. The Society of Cells: Cancer and Control of Cell Proliferation. New York: Springer Verlag; 1999. [Google Scholar]
- 55.Robboy SJ, Welch WR, Young RH, Truslow GY, Herbst AL, Scully RE. Topographic relation of cervical ectropion and vaginal adenosis to clear cell adenocarcinoma. Obstet Gynecol. 1982;60:546–51. [PubMed] [Google Scholar]
- 56.Newbold RR, McLachlan JA. Vaginal adenosis and adenocarcinoma in mice exposed prenatally or neonatally to diethylstilbestrol. Cancer Res. 1982;42:2003–11. [PubMed] [Google Scholar]
- 57.Kurita T, Mills A, Cunha GR. Roles of p63 in the diethylstilbestrol-induced cervicovaginal adenosis. Development. 2004;131:1639–49. doi: 10.1242/dev.01038. [DOI] [PubMed] [Google Scholar]
- 58.Cunha GR. Stromal induction and specification of morphogenesis and cytodifferentiation of the epithelia of the Mullerian ducts and urogenital sinus during development of the uterus and vagina in mice. J Exp Zool. 1976;196:361–70. doi: 10.1002/jez.1401960310. [DOI] [PubMed] [Google Scholar]
- 59.Yin Y, Lin C, Ma L. MSX2 promotes vaginal epithelial differentiation and wolffian duct regression and dampens the vaginal response to diethylstilbestrol. Mol Endocrinol. 2006;20:1535–46. doi: 10.1210/me.2005-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM. Exposure to the xenoestrogen bisphenol-A alters development of the fetal mammary gland. Endocrinology. 2006 doi: 10.1210/en.2006-0561. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Veltmaat JM, Mailleux AA, Thiery JP, Bellusci S. Mouse embryonic mammogenesis as a model for the molecular regulation of pattern formation. Differentiation. 2003;71:1–17. doi: 10.1046/j.1432-0436.2003.700601.x. [DOI] [PubMed] [Google Scholar]
- 62.Weber-Hall SJ, Phippard DJ, Niemeyer CC, Dale TC. Developmental and hormonal regulation of Wnt gene expression in the mouse mammary gland. Differentiation. 1994;57:205–14. doi: 10.1046/j.1432-0436.1994.5730205.x. [DOI] [PubMed] [Google Scholar]
- 63.Phippard DJ, Weber-Hall SJ, Sharpe PT, et al. Regulation of Msx-1, Msx-2, Bmp-2 and Bmp-4 during foetal and postnatal mammary gland development. Development. 1996;122:2729–37. doi: 10.1242/dev.122.9.2729. [DOI] [PubMed] [Google Scholar]
- 64.Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM. Bisphenol A exposure alters fetal mammary gland development; Endocrinology Society Meeting; 2006. pp. 1–177.pp. 205 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic representation of a 5μm mammary gland section. The morphometric analysis was performed in a 4mm2 area that excluded the TEBs. All structures within this area were counted and classified.
Body weights of offspring born to mothers exposed to BPA or vehicle. Mean body weights of male and female offspring (PND 4–11) and mean body weights of female offspring (PND 22–110) showed no statistically significant differences due to BPA treatment.