
Genética médica

| |
| Foco | Condições genéticas e anomalias congênitas |
| Doenças significativas | Embriopatias, erros inatos do metabolismo, displasias esqueléticas, síndromes com malformações congênitas, síndromes neurogenéticas, entre outras |
| Testes significativos | Sequenciamento do exoma, acilcarnitinas, dosagem de aminoácidos, Array CGH, entre outros |
Genética médica ou genética clínica é a especialidade que lida com o diagnóstico, tratamento e controle dos distúrbios genéticos e hereditários. É uma área que enfoca não só o paciente mas também toda a família, principalmente por meio do aconselhamento genético.
Escopo
[editar | editar código-fonte]A Genética Médica compreende diferentes áreas, incluindo o atendimento clínico, a realização de exames de diagnóstico laboratorial e a pesquisa de causas e padrões de herança de doenças genéticas. Exemplos de condições que integram o escopo da Genética Médica incluem os defeitos congênitos e dismorfologia, síndromes teratogênicas, erros inatos do metabolismo, displasias esqueléticas, câncer familial, síndromes neurogenéticas, síndromes hematogenéticas, genodermatoses, entre outras.[1] Há sobreposições crescentes com outras especialidades médicas, uma vez que os avanços recentes da Genética têm revelado a etiologia de condições neurológicas, endócrinas, cardiovasculares, pulmonares, oftalmológicas, renais, psiquiátricas e dermatológicas.
História da Genética Médica
[editar | editar código-fonte]Século XIX e primeira metade do século XX
[editar | editar código-fonte]
Os padrões de herança genética foram primeiramente descritos por Gregor Mendel em 1865 em seus experimentos com ervilhas. Esses conceitos foram aplicados às doenças humanas familiares em 1902, quando Archibald Garrod descreveu o padrão de herança autossômico recessivo para a alcaptonúria. O mesmo pesquisador é responsável pelo termo "erros inatos do metabolismo". As descobertas em relação à herança humana possibilitaram o surgimento das clínicas de aconselhamento genético nos Estados Unidos em 1941.[2] Os atendimentos das primeiras clínicas incluíam temas similares aos contemporâneos como retardo mental, defeitos do tubo neural e doença de Huntington, bem como temas atualmente raramente abordados em contextos clínicos, como cor da pele e dos olhos e gemelaridade.[2]

Segunda metade do século XX até os tempos atuais
[editar | editar código-fonte]Um número importante de progressos na compreensão das bases moleculares da genética ocorreu a partir da segunda metade do século XX. Os cromossomos já haviam sido apontados como base da herança em 1903, mas a estrutura do DNA só seria descrita em 1953 por James Watson e Francis Crick. Em 1956, o número correto de cromossomos foi estabelecido e em 1959, foi identificada a associação entre a síndrome de Down e a trissomia do 21 por Jérôme Lejeune. Um dos mais relevantes trabalhos para a Genética Médica foi a publicação da primeira edição do Mendelian Inheritance in Man (MIM), um catálogo que reuniu todos os genes e doenças genéticas conhecidos, em 1966 por Victor A. McKusick, considerado o pai da medicina genética.[3] O MIM é atualmente disponibilizado de forma gratuita na internet com o nome de OMIM - Online Mendelian Inheritance in Man e é atualizado regularmente. Outros marcos importantes para a Genética Médica incluem a descoberta das endonucleases de restrição em 1970, permitindo a análise do DNA, bem como a publicação do esboço do genoma humano em 2000. Essas descobertas permitiram o diagnóstico de muitas doenças genéticas através da análise cromossômica e de testes moleculares.
Prática clínica atual
[editar | editar código-fonte]As situações clínicas nas quais os pacientes são avaliados determinam o escopo das intervenções práticas, diagnósticas e terapêuticas. De um modo geral, os encontros entre pacientes e médicos geneticistas envolvem:[2]
- Encaminhamentos para ambulatórios de Genética (pediátricos, adultos ou combinados) ou uma interconsulta intra-hospitalar, mais frequentemente para avaliação diagnóstica;
- Ambulatórios de genética especializados, com enfoque no manuseio de erros inatos do metabolismo, displasias esqueléticas ou doenças de depósito lisossômico;
- Encaminhamentos para aconselhamento em ambulatório de genética pré-natal para discutir riscos gestacionais (idade materna avançada, exposição a teratógenos, história familiar de doença genética, história de abortamento de repetição), resultados de testes (alterações ultrassonográficas, testes )de triagem materna alterados e/ou opções de diagnóstico pré-natal (tipicamente amniocentese ou biópsia de vilosidade coriônica);
- Ambulatório multidisciplinares que incluem um médico geneticista (genética do câncer, genética cardiovascular, distúrbios da diferenciação sexual, defeitos craniofaciais ou fendas labiais/palatinas, ambulatórios de surdez, ambulatórios de distrofias musculares/doenças neurodegenerativas).
Aconselhamento Genético
[editar | editar código-fonte]O aconselhamento genético é o processo pelo qual se fornece informação e aconselhamento a pacientes ou familiares, sobre um distúrbio genético herdado que possa afetá-los ou a seus descendentes. O propósito do aconselhamento é auxiliar nas decisões sobre reprodução e outros assuntos relacionados à saúde, com base em informações sobre o distúrbio genético, seu tratamento e prognóstico, os padrões de herança e os testes diagnósticos disponíveis.[4] O termo Aconselhamento Genético também é aplicado à consulta em Genética Médica de um modo geral, visando o diagnóstico, tratamento e prevenção de doenças genéticas.[5]
Avaliação diagnóstica
[editar | editar código-fonte]O diagnóstico em Genética Médica, assim como na Medicina em geral, firma-se principalmente nos pilares da propedêutica: anamnese e exame físico.[5] Entretanto, para alguns casos tornam-se necessários exames complementares. Entre eles, destacam-se, na área específica da Genética, os estudos cromossômicos, metabólicos e moleculares.
Estudos cromossômicos
[editar | editar código-fonte]
Estudos cromossômicos são usados na genética clínica para determinar a causa de atrasos de desenvolvimento, defeitos congênitos, características dismórficas e/ou autismo. A análise cromossômica é também realizada no pré-natal para determinar se um feto é afetado por aneuploidia ou outros rearranjos cromossômicos. Finalmente, as anomalias cromossômicas são frequentemente detectadas em amostras de câncer. Um grande número de diferentes métodos tem sido desenvolvido para análise de cromossomos:
- Análise de cromossomos usando o cariótipo envolve diferentes corantes para gerar bandas claras e escuras, permitindo a identificação de cada cromossomo em um microscópio;
- Cariótipo de instabilidade cromossômica envolve o uso de substâncias que induzem quebras cromossômicas, auxiliando no diagnóstico condições genéticas relacionadas ao reparo do DNA, como a ataxia telangiectasia.
- Hibridação in situ fluorescente (FISH) envolve o uso de sondas marcadas com fluorecência que se ligam a sequencias específicas de DNA, usadas para identificar aneuploidia, deleções genômicas ou duplicações, caracterizando translocações cromossômicas e determinando a origem de cromossomos em anel;
- Hibridação genômica comparativa em array (Array CGH) é uma técnica molecular nova que envolve a hibridização de uma amostra de DNA individual a uma lâmina de vidro ou chip de microarranjos contendo sondas moleculares que representam regiões únicas do genoma. Esse método é particularmente sensível para detecção de ganhos ou perdas cromossômicas ao longo do genoma, mas não detecta translocações balanceadas ou distingue o local da duplicação do material genético.[6]
Estudos metabólicos básicos
[editar | editar código-fonte]Estudos bioquímicos são realizados para rastrear por alterações nos metabólitos em fluidos corporais, geralmente o sangue ou urina, mas também ocasionalmente o líquido cefalorraquidiano. Testes específicos de função enzimática (em leucócitos, fibroblastos da pele, fígado ou músculo) também são utilizados em determinadas circunstâncias:
- A dosagem quantitativa de aminoácidos é tipicamente realizada utilizando a reação de ninidrina, seguida de cromatografia líquida para medir a quantidade de aminoácidos na amostra (urina, plasma/soro ou líquor). A medida de aminoácidos no plasma ou soro é utilizada na avaliação de doenças do metabolismo de aminoácidos como transtornos do ciclo da ureia, doença da urina em xarope de bordo e fenilcetonúria. As medidas de aminoácidos na urina podem ser úteis no diagnóstico de cistinúria ou síndrome de Fanconi renal como pode ser visto na cistinose;
- A análise de ácidos orgânicos na urina pode ser realizada tanto por métodos qualitativos como quantitativos, mas em todo caso o teste é usado para detetar a excreção de ácidos orgânicos anormais. Esses compostos são normalmente produzidos durante o metabolismo corporal de aminoácidos e ácidos graxos de cadeia ímpar, mas acumulam-se em pacientes com certas doenças metabólicas;
- O perfil de acilcarnitinas deteta compostos como os ácidos orgânicos e ácidos graxos conjugados a carnitina. O teste é utilizado para deteção de doenças envolvendo o metabolismo de ácidos graxos.
- Piruvato e lactato são produtos do metabolismo normal, particularmente durante o metabolismo anaeróbico. Esses compostos normalmente se acumulam durante exercício ou isquemia, mas estão também elevados em pacientes com transtornos do metabolismo do piruvato ou doenças mitocondriais;
- Amônia é um produto final do metabolismo dos aminoácidos e é convertida no fígado em ureia por uma série de reações enzimáticas conhecidas como ciclo da ureia. Amônia aumentada pode então ser detetada em pacientes com transtornos do ciclo da ureia, bem como outras condições incluindo insuficiência hepática;
- Testes enzimáticos são realizados para uma grande quantidade de doenças metabólicas para confirmar o diagnóstico suspeitado pelos testes de triagem.
Estudos moleculares
[editar | editar código-fonte]- O sequenciamento do DNA é utilizado para analisar diretamente a sequência de DNA genômico de um gene em particular. Em geral, apenas as partes do gene que codificam a proteína expressa (éxons) e pequenas quantidades das regiões não traduzidas e íntrons são analisados. Desta forma, embora os testes sejam altamente específicos e sensíveis, eles não rotineiramente identificam todas as mutações que podem causar a doença.
- A análise de metilação do DNA é utilizada para diagnosticar certas doenças genéticas que são causadas por mecanismos epigenéticos como o imprinting genômico e a dissomia uniparental.
- A análise de DNA pela técnica de Southern Blot é útil no diagnóstico de doenças causadas por repetições de trinucleotídeos. Outra técnica utilizada nesses casos é a Triplet repeat primed PCR (TP PCR).[7]
Tratamento
[editar | editar código-fonte]Cada célula do corpo contém informação hereditária (ADN) envolta em estruturas chamadas cromossomos. Uma vez que as síndromes genéticas tipicamente resultam de alterações nos cromossomos ou genes, não há tratamento disponível atualmente que possa corrigir as alterações genéticas. Assim, ainda não há "cura" para doenças genéticas. Entretanto, em alguns casos, particularmente nos erros inatos do metabolismo, o mecanismo da doença é bem entendido e oferece o potencial de manuseio dietético e medicamentoso para prevenir ou reduzir as complicações no longo prazo. Em outros casos, a terapia de infusão é usada para repor a enzima deficiente. As pesquisas atuais estão buscando ativamente o desenvolvimento de novas medicações para tratar doenças genéticas específicas.
Manuseio dietético
[editar | editar código-fonte]
Restrição e suplementação dietética são medidas tomadas em diversas doenças metabólicas bem conhecidas, incluindo galactosemia, intolerância hereditária a Frutose, fenilcetonúria, doença da urina em xarope de bordo, acidúrias orgânicas e doenças do ciclo da ureia. Tais dietas restritivas podem ser difíceis de serem mantidas pelo paciente e sua família e requer acompanhamento com nutricionista com experiência em doenças metabólicas. A composição da dieta muda de acordo com as necessidades calóricas da criança em crescimento e atenção especial é necessária durante a gravidez se uma mulher é afetada por uma dessas doenças.
Moléculas pequenas
[editar | editar código-fonte]- Reposição do produto enzimático ou outras substancias deficientes A falta de um produto enzimático pode ser a principal causa dos sintomas apresentados em certas doenças metabólicas, que podem então ser controladas com a reposição desse produto. Exemplos incluem a reposição de levodopa nas distonias responsivas a levodopa [8] e administração de biotina na deficiência de biotinidase.[9] Em algumas doenças relacionadas a proteínas transportadoras, a suplementação da substância deficiente também é efetiva no tratamento. Exemplos incluem a suplementação de vitamina E na ataxia com deficiência de vitamina E[8] e de cobre na doença de Menkes.[10]
- Terapia de redução do substrato Em algumas doenças relacionadas a defeitos enzimáticos, como certas doenças de depósito lisossômico, os sintomas decorrem principalmente do acúmulo do substrato e a inibição de sua síntese é utilizada para fins terapêuticos. Exemplos incluem o uso do eliglustate na Doença de Gaucher[11] e da genisteína nas mucopolissacaridoses.[12]
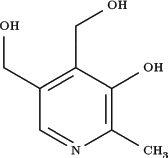
Estrutura da piridoxina, ou vitamina B6, utilizada no tratamento da homocistinúria clássica responsiva a piridoxina e na epilepsia dependente de piridoxina.

- Cofatores e moléculas potencializadoras Em algumas doenças metabólicas, a atividade enzimática residual pode ser acentuada com o uso de vitaminas ou cofatores relacionados ao funcionamento da enzima. Um exemplo é o uso de altas doses de piridoxina em alguns pacientes com homocistinúria para aumentar a atividade residual da enzima cistationa sintase.[13] Além disso, em certas condições não relacionadas a um defeito enzimático, a função da proteína defeituosa pode ser potencializada por certas substancias, como no uso do ivacaftor para potencializar a proteína CFTR em um subgrupo de pacientes com fibrose cística.[14]
- Chaperonas farmacológicas Alguns pacientes com doenças genéticas tem mutações de sentido trocado, que provocam uma alteração na conformação da proteína codificada, provocando sua degradação. Certos medicamentos atuam como chaperonas farmacológicas, estabilizando a proteína e aumentando a quantidade de proteína funcional. Exemplos incluem o uso do lumacaftor na fibrose cística [15] e do migalastate na doença de Fabry [16].
- Read-through Uma certa parcela dos pacientes com doenças genéticas possuem mutações sem sentido, que levam a um códon de parada prematuro e à degradação do RNA mensageiro. Certas moléculas pequenas que permitem a tradução do RNA mensageiro através desses códons (read-through). Em maio de 2014, um representante dessa classe de medicamentos, o atalureno, recebeu aprovação condicional pela Agência Europeia de Medicamentos (EMA) para o tratamento de pacientes com Distrofia muscular de Duchenne com mutações sem sentido.[17]
- Outras moléculas pequenas farmacêuticas Certas doenças genéticas envolvem proteínas que possuem um papel bem conhecido, possibilitando a atuação nas vias envolvidas na patogênese. Exemplos incluem o uso do pamidronato para reduzir a reabsorção óssea na osteogênese imperfeita,[18] do tafamidis e diflunisal para estabilizar os tetrâmeros de transtirretina na polineuropatia amiloidótica familiar.[19]
Medicamentos biológicos e oligonucleotídios
[editar | editar código-fonte]- Reposição enzimática ou de outras proteínas Certas doenças de depósito lisossômico são tratadas com infusão de uma enzima recombinante (produzida em laboratório) que pode reduzir o acúmulo de compostos em diversos tecidos. Exemplos incluem a doença de Gaucher,[20] a doença de Fabry,[21] as mucopolissacaridoses[22] e a doença de Pompe.[23] Em outros casos, o tratamento consiste na reposição de proteínas não enzimáticas, como a reposição de fatores de coagulação nas coagulopatias hereditárias.[24] e de hormônio de crescimento na deficiência de hormônio de crescimento.[25]
- Terapia gênica A terapia gênica compreende o uso de genes com fins terapêuticos[26] Estudos clínicos com a terapia gênica tiveram início no começo da década de 1990 e a tecnologia vem sendo aperfeiçoada desde então. Em novembro de 2012, o primeiro produto farmacológico para terapia gênica para uma doença monogênica, o alipogene tipavorvec, usado para o tratamento da deficiência de lipoproteína lipase, foi aprovado pela EMA.[27] Por razões comerciais, o medicamento foi descontinuado em 2017, após ter sido utilizado comercialmente uma única vez.[28] Atualmente, a terapia gênica é utilizada comercialmente para o tratamento da imunodeficiência severa combinada por deficiência de adenosina deaminase[29] e para a atrofia muscular espinhal[30] e há diversos outros produtos em fases finais de desenvolvimento.[28]
- Terapia antisenso Oligonucleotídios antisenso podem ser utilizados para inibir ou modificar a tradução do RNA mensageiro em certas condições genéticas. Exemplos incluem o uso do eteplirseno na distrofia muscular de Duchenne para "saltar" o éxon 51 do gene DMD, durante o processo de encadeamento do RNA mensageiro, resultando em uma proteína menor, porém funcional (exon skipping)[31] e da nusinersena na atrofia muscular espinhal para aumentar a produção da proteína SMN de tamanho inteiro a partir do gene SMN2.[32]
- RNA interferente O RNA interferente (RNAi) compreende o uso de conjugados oligonucleotídicos modificados para silenciar o RNA mensageiro através do complexo de silenciamento induzido por RNA (RISC). Exemplos incluem o uso da patisirana, desenvolvida para o tratamento da polineuropatia amiloidótica familiar [33], e da givosirana, para as porfirias hepáticas agudas.
Transplantes
[editar | editar código-fonte]Uma parcela das doenças genéticas decorre da mutação de proteínas que são normalmente produzidas totalmente ou principalmente em órgãos específicos, que podem ser transplantados. Além disso, o transplante de medula óssea é usado para suplementar enzimas e outros produtos gênicos ausentes em determinadas doenças. Exemplos incluem o transplante hepático na polineuropatia amiloidótica familiar [19] e o transplante de medula óssea na Adrenoleucodistrofia ligada ao X e em certas doenças de depósito lisossômico.[34]
Carreira em Genética Médica
[editar | editar código-fonte]A Genética Médica, vista como sinônimo de Genética Clínica, é uma especialidade médica. Para a exercer, é necessário graduar-se em uma escola médica e especializar-se em Genética Médica. No Brasil, esse treinamento leva nove anos, incluindo seis anos de Medicina e três anos de Genética Médica. O termo Medical Genetics é utilizado em sentido mais amplo nos Estados Unidos e outros países, incluindo não só a Genética Clínica (Clinical Genetics), como também a Genética Clínica Bioquímica (Clinical Biochemical Genetics), a Citogenética Clínica (Clinical Cytogenetics) e a Genética Clínica Molecular (Clinical Molecular Genetics).[35] As demais áreas são campos de atuação comuns com outros profissionais de saúde.
Genética Médica no Brasil
[editar | editar código-fonte]A primeira residência em Genética Médica no Brasil foi criada em 1977 no Hospital das Clínicas de Ribeirão Preto da Universidade de São Paulo. O reconhecimento como especialidade médica pelo Conselho Federal de Medicina ocorreu apenas seis anos depois, em 1983, através da resolução Nº 1.128/1983.[36] Em 1986, foi fundada a Sociedade Brasileira de Genética Médica e Genômica - SBGM, a partir da iniciativa dos membros da Sociedade Brasileira de Genética.[37] Em pesquisa da demografia médica em 2023, havia 407 médicos geneticistas, sendo a especialidade médica com menos integrantes.[38]
Matriz de competências dos programas de residência médica
[editar | editar código-fonte]O conteúdo e enfoque dos programas de residência médica em genética médica no Brasil é planejado de modo a permitir que os residentes atinjam as competências determinadas na resolução nº 20, de 8 de abril de 2019.[39] As competências necessárias ao término do treinamento incluem habilidades e atitudes relacionadas a investigação clínica, investigação laboratorial e manuseio de condições genéticas, bem como a comunicação e relacionamento com pacientes e familiares, além de aspectos relacionados à saúde coletiva e à gestão do conhecimento.
Genética Médica no SUS
[editar | editar código-fonte]Apesar de alguns serviços de saúde públicos oferecerem atendimento em Genética Médica, não há uma rede de atenção integrada e de acesso universal nessa área.[40] Essa situação começou a ser solucionada em 2004, com a publicação da portaria Nº 2.380,[41] que instituiu o grupo de trabalho de Genética Clínica, com a finalidade de elaborar a Política Nacional de Atenção à Saúde em Genética Clínica. Tal tarefa foi concluída em 2009, quando foi publicada a portaria Nº 81.[42] Em 2012, entretanto, foi instituído um novo grupo de trabalho para formulação da Política de Atenção às Pessoas com Doenças Raras, com o objetivo de regulamentar a atenção não só aos pacientes com doenças genéticas mas também aos com outras doença raras. As diretrizes assistenciais e as normas para habilitação de hospitais e serviços para o atendimento foram publicadas em janeiro de 2014.[43] Os primeiros serviços de referência em doenças raras foram credenciados em 2016 e há atualmente oito serviços habilitados no país.[44]
Genética Médica em Portugal e na União Europeia
[editar | editar código-fonte]Em Portugal, a genética médica foi primeiramente reconhecida como uma competência em 1983. Para o acesso à competência de Genética Médica, era necessário ter a especialidade de Pediatria, Medicina Interna, Neurologia ou Endocrinologia. Em 1998, foi aprovada em reunião do Conselho Nacional Executivo da Ordem dos Médicos a passagem de competência a especialidade e, em 2001, foi criada a área profissional de Genética Médica e publicado o respectivo programa de formação do internato complementar.[45] Em 2011, havia 24 especialistas em genética médica em Portugal.[46]
Na União Europeia, a Genética Médica foi oficialmente reconhecida como especialidade apenas em 2011.[47] Ainda em 2024, a Genética Médica não é reconhecida oficialmente na Espanha, sendo o único país da União Europeia sem a regulamentação da especialidade.[48]
Referências
- ↑ «Proposição de Conteúdo dos Programas de Residência Médica». Consultado em 27 de outubro de 2012
- ↑ a b c Kingston, HM. (2001). ABC of clinical genetics. Londres: BMJ. ISBN 9780727916273
- ↑ «Profiles in Science: The Victor A. McKusick Papers». Consultado em 27 de outubro de 2012
- ↑ «DeCS - Descritores em Ciências da Saúde». Consultado em 27 de outubro de 2012
- ↑ a b Brunoni, Décio (2002). «Aconselhamento Genético». Ciência & Saúde Coletiva. 7 (1): 101-107. ISSN 1413-8123. doi:10.1590/S1413-81232002000100009. Consultado em 27 de outubro de 2012
- ↑ Rickman, L (9 de setembro de 2005). «Prenatal detection of unbalanced chromosomal rearrangements by array CGH». Journal of Medical Genetics. 43 (4): 353-361. ISSN 1468-6244. doi:10.1136/jmg.2005.037648. Consultado em 27 de outubro de 2012
- ↑ Warner, J P; L H Barron; D Goudie; K Kelly; D Dow; D R Fitzpatrick; D J Brock (1 de dezembro de 1996). «A general method for the detection of large CAG repeat expansions by fluorescent PCR». Journal of Medical Genetics. 33 (12): 1022–1026. ISSN 1468-6244. doi:10.1136/jmg.33.12.1022. Consultado em 9 de novembro de 2014
- ↑ a b Jinnah, H. A.; Albanese, Alberto; Bhatia, Kailash P.; Cardoso, Francisco; Da Prat, Gustavo; de Koning, Tom J.; Espay, Alberto J.; Fung, Victor; Garcia-Ruiz, Pedro J. «Treatable inherited rare movement disorders». Movement Disorders (em inglês): n/a–n/a. ISSN 1531-8257. doi:10.1002/mds.27140
- ↑ Zempleni Janos; Yousef I Hassan; Subhashinee SK Wijeratne (novembro de 2008). «Biotin and biotinidase deficiency». Expert Review of Endocrinology & Metabolism. 3 (6): 715-724. ISSN 1744-6651. doi:10.1586/17446651.3.6.715. Consultado em 27 de outubro de 2012
- ↑ Kaler, Stephen G.; Liew, Clarissa J.; Donsante, Anthony; Hicks, Julia D.; Sato, Susumu; Greenfield, Jacquelyn C. (1 de outubro de 2010). «Molecular correlates of epilepsy in early diagnosed and treated Menkes disease». Journal of Inherited Metabolic Disease (em inglês). 33 (5): 583–589. ISSN 0141-8955. doi:10.1007/s10545-010-9118-2
- ↑ Mistry, Pramod K.; Lukina, Elena; Ben Turkia, Hadhami; Shankar, Suma P.; Baris, Hagit; Ghosn, Marwan; Mehta, Atul; Packman, Seymour; Pastores, Gregory (1 de novembro de 2017). «Outcomes after 18 months of eliglustat therapy in treatment-naïve adults with Gaucher disease type 1: The phase 3 ENGAGE trial». American Journal of Hematology (em inglês). 92 (11): 1170–1176. ISSN 1096-8652. doi:10.1002/ajh.24877
- ↑ Coutinho, Maria Francisca; Santos, Juliana Inês; Alves, Sandra (4 de julho de 2016). «Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders». International Journal of Molecular Sciences (em inglês). 17 (7). 1065 páginas. doi:10.3390/ijms17071065
- ↑ Skovby Flemming; Mette Gaustadnes; S. Harvey Mudd (janeiro de 2010). «A revisit to the natural history of homocystinuria due to cystathionine β-synthase deficiency». Molecular Genetics and Metabolism. 99 (1): 1-3. ISSN 1096-7192. doi:10.1016/j.ymgme.2009.09.009. Consultado em 27 de outubro de 2012
- ↑ De Boeck Kris; Anne Munck; Seth Walker; Albert Faro; Peter Hiatt; Geoffrey Gilmartin; Mark Higgins (26 de setembro de 2014). «Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation». Journal of Cystic Fibrosis: Official Journal of the European Cystic Fibrosis Society. ISSN 1873-5010. PMID 25266159. doi:10.1016/j.jcf.2014.09.005
- ↑ Cutting, Garry R. (5 de dezembro de 2017). «Treating Specific Variants Causing Cystic Fibrosis». JAMA. 318 (21): 2130–2131. ISSN 0098-7484. doi:10.1001/jama.2017.16823
- ↑ Germain, Dominique P.; Hughes, Derralynn A.; Nicholls, Kathleen; Bichet, Daniel G.; Giugliani, Roberto; Wilcox, William R.; Feliciani, Claudio; Shankar, Suma P.; Ezgu, Fatih (10 de agosto de 2016). «Treatment of Fabry's Disease with the Pharmacologic Chaperone Migalastat». New England Journal of Medicine (em inglês). 375 (6): 545–555. doi:10.1056/nejmoa1510198
- ↑ European Medicines Agency (23 de maio de 2014). «European Medicines Agency recommends first-in-class medicine for treatment of Duchenne muscular dystrophy» (PDF)
- ↑ Brasil. «PORTARIA Nº 1.306, DE 22 DE NOVEMBRO DE 2013». Diário Oficial da União 228 ed. Brasília. pp. 58–60
- ↑ a b Ueda, Mitsuharu; Yukio, Ando (2014). «Recent advances in transthyretin amyloidosis therapy». Translational Neurodegeneration. 3. 19 páginas. ISSN 2047-9158. PMC 4165622
 . PMID 25228988. doi:10.1186/2047-9158-3-19
. PMID 25228988. doi:10.1186/2047-9158-3-19
- ↑ Martins Ana M.; Clarisse L. Lobo; Elisa A. P. Sobreira; Eugenia R. Valadares; Gilda Porta; José Semionato Filho; Mara A. D. Pianovsky; Marcelo S. Kerstenetzky; Maria F. P. Montoril; Paulo C. Aranda; Ricardo F. Pires; Ronald M. V. Mota; Teresa C. Bortolheiro; Maria T. M. Paula (junho de 2003). «Tratamento da doença de Gaucher: um consenso brasileiro». Revista Brasileira de Hematologia e Hemoterapia. 25 (2). ISSN 1516-8484. doi:10.1590/S1516-84842003000200004. Consultado em 27 de outubro de 2012
- ↑ Schiffmann, R. (6 de junho de 2001). «Enzyme Replacement Therapy in Fabry Disease: A Randomized Controlled Trial». JAMA: The Journal of the American Medical Association. 285 (21): 2743-2749. ISSN 0098-7484. doi:10.1001/jama.285.21.2743. Consultado em 27 de outubro de 2012
- ↑ Giugliani Roberto et al. (2010). «Terapia de reposição enzimática para as mucopolissacaridoses I, II e VI: recomendações de um grupo de especialistas brasileiros». Revista da Associação Médica Brasileira. 56 (3): 271-277. ISSN 0104-4230. doi:10.1590/S0104-42302010000300009. Consultado em 27 de outubro de 2012
- ↑ van der Beek; M L C Hagemans; A T van der Ploeg; A J J Reuser; P A van Doorn (junho de 2006). «Pompe disease (glycogen storage disease type II): clinical features and enzyme replacement therapy». Acta neurologica Belgica. 106 (2): 82-8. ISSN 0300-9009. Consultado em 27 de outubro de 2012
- ↑ MINISTÉRIO DA SAÚDE (2002). Manual de tratamento das Coagulopatias Hereditárias. [S.l.: s.n.] ISBN 85-334-0993-1
- ↑ Hardin, DS (2008). «Treatment of short stature and growth hormone deficiency in children with somatotropin (rDNA origin)». Biologics
- ↑ Wang, Dan; Guangping, Gao (setembro de 2014). «State-of-the-art human gene therapy: part II. Gene therapy strategies and clinical applications». Discovery Medicine. 18 (98): 151–161. ISSN 1944-7930. PMID 25227756
- ↑ «Gene therapy: Glybera approved by European Commission». Consultado em 27 de janeiro de 2013
- ↑ a b Senior, Melanie (7 de junho de 2017). «After Glybera's withdrawal, what's next for gene therapy?». Nature Biotechnology (em inglês). 35 (6): 491–492. doi:10.1038/nbt0617-491
- ↑ Anonymous (17 de setembro de 2018). «Strimvelis». European Medicines Agency (em inglês). Consultado em 24 de junho de 2019
- ↑ Commissioner, Office of the (24 de maio de 2019). «FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality». FDA (em inglês). Consultado em 24 de junho de 2019
- ↑ Lim, Kenji Rowel; Maruyama, Rika; Yokota, Toshifumi (28 de fevereiro de 2017). «Eteplirsen in the treatment of Duchenne muscular dystrophy». Drug Design, Development and Therapy (em inglês). Volume11: 533–545. doi:10.2147/dddt.s97635
- ↑ Finkel, Richard S.; Mercuri, Eugenio; Darras, Basil T.; Connolly, Anne M.; Kuntz, Nancy L.; Kirschner, Janbernd; Chiriboga, Claudia A.; Saito, Kayoko; Servais, Laurent (1 de novembro de 2017). «Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy». New England Journal of Medicine (em inglês). 377 (18): 1723–1732. doi:10.1056/nejmoa1702752
- ↑ Hoy, Sheridan M. (1 de outubro de 2018). «Patisiran: First Global Approval». Drugs (em inglês). 78 (15): 1625–1631. ISSN 1179-1950. doi:10.1007/s40265-018-0983-6
- ↑ Lange Marcos C.; Hélio A.G. Teive; André R. Troiano; Marco Bitencourt; Vaneuza A.M. Funke; Daniela C. Setúbal; José Zanis Neto; Carlos R. Medeiros; Lineu C. Werneck; Ricardo Pasquini; Carmen M.S. Bonfim (março de 2006). «Bone marrow transplantation in patients with storage diseases: a developing country experience». Arquivos de Neuro-Psiquiatria. 64 (1): 1–4. ISSN 0004-282X. doi:10.1590/S0004-282X2006000100001. Consultado em 8 de novembro de 2014
- ↑ «Specialties of Genetics». ABMG. Consultado em 27 de outubro de 2012. Arquivado do original em 1 de janeiro de 2010
- ↑ «Resolução CFM Nº 1128/1983». Consultado em 27 de outubro de 2012
- ↑ «Histórico da Sociedade Brasileira de Genética Médica». Consultado em 27 de outubro de 2012
- ↑ Scheffer, M (2023). Demografia Médica no Brasil 2023. São Paulo: Conselho Regional de Medicina do Estado de São Paulo e Conselho Federal de Medicina. 346 páginas
- ↑ «RESOLUÇÃO Nº 20, DE 8 DE ABRIL DE 2019 - Imprensa Nacional». Consultado em 6 de dezembro de 2019
- ↑ Novoa, Maria Concepción; Burnham, Teresinha Fróes (janeiro de 2011). «Desafios para a universalização da genética clínica: o caso brasileiro». Revista Panamericana de Salud Pública. 29 (1): 61-68. ISSN 1020-4989. doi:10.1590/S1020-49892011000100010. Consultado em 27 de outubro de 2012
- ↑ «PORTARIA Nº 2380/GM». Consultado em 28 de outubro de 2023
- ↑ «PORTARIA Nº 81 DE 20 DE JANEIRO DE 2009». Consultado em 28 de outubro de 2023
- ↑ Brasil. «PORTARIA Nº 199, DE 30 DE JANEIRO DE 2014». Diário Oficial da União 30 ed. Brasília. pp. 44–54
- ↑ «SBGM - Sociedade Brasileira de Genética Médica». www.sbgm.org.br. Consultado em 24 de junho de 2019
- ↑ «Colégio da Especialidade de Genética Médica – Ordem dos Médicos». Consultado em 24 de junho de 2019
- ↑ Peixoto, H (2013). Estudo de Evolução Prospectiva de Médicos no Sistema Nacional de Saúde. Coimbra: Universidade de Coimbra. 162 páginas
- ↑ Kristoffersson, Ulf; Macek, Milan (2017). «From Mendel to Medical Genetics». European Journal of Human Genetics (em inglês). 25 (S2): S53–S5 9. ISSN 1018-4813. doi:10.1038/ejhg.2017.157
- ↑ Linde, Pablo (16 de março de 2024). «Años de espera y gastos para un diagnóstico: España es el único país de la UE sin especialidad de genética clínica». El País (em espanhol). Consultado em 1 de junho de 2024